




INTRODUCTION
Compared with monogastric animals, the ruminants have a more specialized stomach composed of four separate chambers and the rumen is the largest. In the rumen, there are high-density microorganisms, such as bacteria, archaea, fungi, and protozoa. These microorganisms play a vital role in maintaining the normal digestion and absorption in the host. They can digest the fibers into monosaccharides and disaccharides and then convert them into fatty acids, which can be absorbed and used by the host [1,2].
It was previously reported that ruminal Prevotella in sheep was more diverse with the increase of fiber content in ration [3], the abundance of Bacteroides in the rumen of steers decreased with the increase of neutral detergent fiber in diets [4], and the rumen Fibrobacteraceae concentration was higher in cows fed with total mixed ration than the free-range cows [5]. These previous researches suggested that diets had significant impact on ruminal microbial community. However, more experimental evidence is needed to confirm this postulation. Moreover, many kinds of ruminants including cows, cattle, buffalo, yak and sheep have been used as experimental subjects to determine the microbial diversity in rumen [2,6,7], while goats are rarely used in studies. And, the previous goat-related studies [8,9] mainly used conventional methods, such as molecular fingerprint, to evaluate the ruminal microbial community. These methods could only identify the dominant species instead of the complete composition of the microorganisms in sample when microbial diversity was investigated [10]. Therefore, the impact of changes in diets on the microflora in goat still remains unclear. High-throughput sequencing is a novel technology, which can process millions of sequenced reads at the same time and obtain the biological information of most microorganisms in sample through deep sequencing, has been recently applied in microbial ecology [11]. In the present study, high-throughput sequencing was applied to investigate the impact of the difference in diet on the microbial structure and composition of the rumen in goats.
MATERIALS AND METHODS
The animal experimental procedures were approved by the Committee of Laboratory Animal Welfare and Ethics of the Sichuan Agricultural University.
Animal experiment and rumen sampling
Twelve castrated 1-yr-old hybrid goats (Jianchang black goat and Boer goat crossbred) with an average body weight of 25┬▒2 kg were used in this study. They were divided into two groups (6 goats per group) according to their age and weight and assigned to two treatments, CF group offered complete feed and AF group offered all-forage diet. The chemical composition of diets for CF and AF are shown in Supplementary Table S1a and S1b, respectively. The goats were kept in separate cages with free access to water. They were fed a restricted dry matter at the level of 3% of their body in equal amounts at 09:00 and 17:00 h. After 20 days of adaption, rumen contents (50 mL) were collected from each goat using a stomach tube before morning feeding. The rumen contents were strained by four layers of gauze and the rumen fluid was aliquoted into 10 mL centrifuge tubes. The samples were sealed and stored at ŌłÆ80┬░C until DNA extraction.
Polymerase chain reaction amplification and sequencing
Total DNA of samples was extracted using DNA Stool Kit (TIANGEN, Beijing, China). The universal prokaryote primer set, 515F and 806R [12], were used to amplify the V4 region of 16S rRNA. Three replicate polymerase chain reactions (PCRs) were performed for each DNA sample. The PCR reaction volume was 20 ╬╝L, including 0.4 ╬╝L FastPfu Polymerase (Transgen, Bio Inc., Beijing, China), 4 ╬╝L 5├ŚFastPfu buffer, 0.4 ╬╝L of each forward and reverse primers (5 ╬╝M), 2 ╬╝L deoxynucleoside triphosphates mix (dNTP, 2.5 mM) and template, DNA 10 ng. The ddH2O was added to a final volume of 20 ╬╝L. The PCR was performed using a thermal cycler Model (ABI GeneAmp 9700, Applied Biosystems Inc., San Diego, CA, USA) with the thermal cycling conditions: initial denaturation of 2 mi at 95┬░C, 30 cycles of denaturation at 95┬░C for 30 s, annealing at 56┬░C for 30 s, extension at 72┬░C for 30 s, final extension at 72┬░C for 5 min and hold at 10┬░C. The PCR products from the same sample were mixed together and purified using the AxyPrep DNA Gel Extraction kit (AXYGEN, Silicon Valley, MA, USA). Purified amplicons were paired-end sequenced (2├Ś150 nt reads) on Illumina MiSeq platform at the MaJorbio Co. of ShangHai (ShangHai, China), as described before [13].
Sequence analysis
The primer sequences and multiplexing barcodes were trimmed. The poor/low quality sequences including those with uncertain nucleotides, continuous three nucleotides with Q value less than 20, unmatched barcode sequences were discarded. The obtained sequences were clustered using Uparse7.0 into operational taxomonic units (OTUs) based on 97% similarity. Chimeras were removed using UCHIME 4.2.4 [14]. The most abundant sequence was selected as the representative for each OTU, and then aligned against the Greengenes database (http://greengenes.lbl.gov) using PyNAS [15]. Representative sequences were taxonomically classified using the ribosomal database project (RDP) taxonomic database. Calculations for rarefraction curves, the estimated maximum number of OTUs, Shannon-wiener index and chaoI index were performed using QIIME version 1.8.0 [16]. The current % coverage was calculated by dividing the observed number of OTUs by the maximum number of OTUs estimated. The Weighted UniFrac distance metric was calculated to compare the phylogenetic similarity between the bacterial communities. Principal co-ordinate analysis (PCoA) cluster plot was performed using R version 3.1.0 for visualization. All Illumina sequences data in this study were deposited to the SRA of the NCBI database under BioProject PRJNA290546.
Statistical analysis
The relative abundance of microbes was log-transformed [17] and the unpaired two-tailed t-test was performed using the SPSS Statistics software v. 19.0 (IBM, Armonk, NY, USA) to assess whether there was a significant difference between CF and AF. Significance level was set at p<0.05.
RESULTS
Data depth and alpha diversity
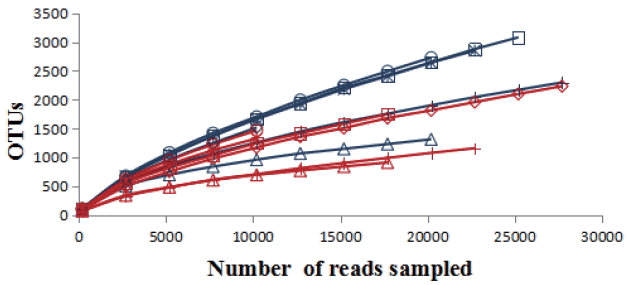
A total of 251,229 sequences were generated after quality control with a mean of 20,809┬▒6,497 (mean┬▒ standard deviation [SD], n = 12) per sample. These sequences were totally clustered into 13,615 OTUs with a mean of 1,983┬▒768 (mean┬▒SD, n = 12) per sample. There were 6,242 OTUs in CF group and 9,510 OTUs in AF group. The difference in the average number of OTUs between the two groups was not significant (p>0.05) (Supplementary Figure S1). The rarefaction curve (Figure 1) showed that most samples were nearly asymptotic, which indicated that the depth of sequencing in the present study could cover most of the microorganisms in the sample. The average of the estimated maximum number of OTUs and current % coverage of each sample at 97% similarity is 3,376 and 65.3%, respectively. Alpha diversity is presented in Table 1. Shannon-Wiener index, ChaoI index and the observed-species were all higher in CF than that in AF. However, the differences were not statistically significant (p>0.05).
Rumen bacterial composition
The sequences in the present experiment were finally annotated as bacteria (99.11%┬▒40.25%), archaea (0.20%┬▒0.23%) and the unassigned microorganisms (0.69%┬▒0.39%). The bacteria kingdom is composed of 23 phyla, of which 22 were identified in CF group and 19 in AF group. The phyla with a relative abundance greater than 1% were Bacteroides, Firmicutes, Proteobacteria, Tenericutes, Verrucomicrobia, and Lentisphaerae in CF, Bacteroides, Firmicutes, Proteobacteria, and Verrucomicrobia in AF. The abundance of Firmicutes in CF was significantly lower (p = 0.027) than that in AF. In contrast, the abundance of Proteobacteria was extremely significantly lower (p<0.001) in AF than that in CF. There was no significant difference in the abundance of other phyla in the two groups (Table 2). Moreover, the bacterial sequences not affiliated with any known phylum were annotated as unclassified bacteria, which made up 2.99%┬▒2.67% and 3.56%┬▒4.07% of the total microbiome in CF and AF group, respectively. At genus level, there were 15 members which differed significantly (p<0.05) in abundance between CF and AF (Table 3). They mainly came from the phylums Firmicutes and Proteobacteria.
Shared genera
There were 33 shared genera within the CF, 30 within the AF and 26 shared by both groups (Table 4). Eight shared genera in CF were greater than 1% in abundance, including Prevotella, Pseudomonas, Subdivision5 genera incertae sedis, Ruminococcus, Butyrivibrio, Succiniclasticum, Clostridium IV, and Vampirovibrio. Seven shared genera in AF were greater than 1% in abundance, including Prevotella, Ruminococcus, Lachnospiracea incertae sedis, Butyrivibrio, Succiniclasticum, Saccharofermentans, and Subdivision5 genera incertae sedis. There were eight genera shared by both groups with the average abundance greater than 1%, including Prevotella, Ruminococcus, Butyrivibrio, Succiniclasticum, Clostridium IV, Lachnospiracea incertae sedis, Saccharofermentans, and Subdivision5 genera incertae sedis.
Weighted unifrac distance analysis
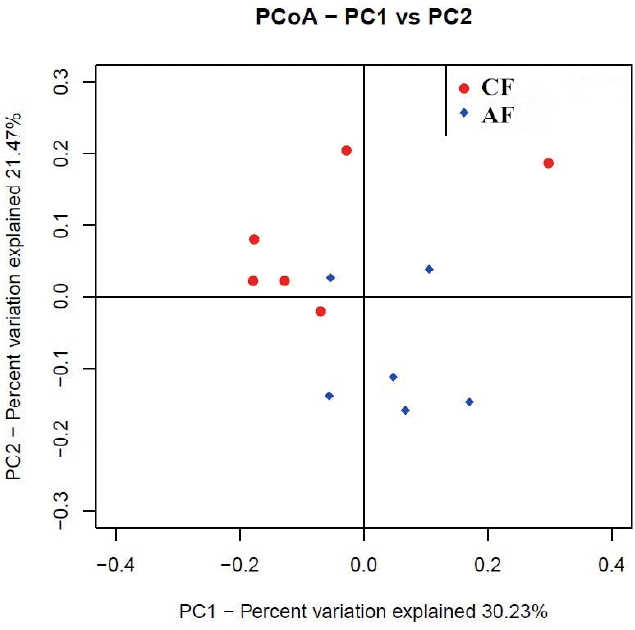
The weighted uniFrac distance matrices were calculated according to the types and abundance of OTU of each sample. Similarity analysis based on the distance matrices showed that the similarity between CF and AF was 61.99%. The intra-group similarity of CF and AF was 62.76% and 67.94%, respectively. The PCoA plot (Figure 2) showed that four samples in CF clustered together while the remaining two were far from them, and samples in AF were evenly distributed in the cluster. All samples tended to cluster together in accordance with their own ration treatment.
DISCUSSION
In this study, the predominant phyla in the rumen of goats were Bacteroidetes and Firmicutes (Table 2), which accounted for 80.3% and 90.7% of the total microbial abundance in CF and AF, respectively. This was consistent with many past studies in herbivores [18ŌĆō20]. At phylum level, Firmicutes had significantly lower abundance in CF (25.87%) than that in AF (42.80%). We explored the reasons at a deeper classification and discovered that four genera within phylum Firmicutes had significantly lower abundance in CF than that in AF, and 3 of them including Lachnospiracea incertae sedis, (phylum) Firmicutes and Ruminococcus were worth of noting because of their high relative abundance. These differences in abundance might due to the different fiber content in diets (Supplementary Table S1). Ruminococcus is composed of two strong fiber-digesting bacteria species, Ruminococcus albus and Ruminococcus flavefaciens, which can produce a large amount of Cellulases and hemicellulases [21]. The higher abundance of Ruminococcus corresponding to the higher fiber content probably came from the effect of substrate-induced. It was reported that rumen Lachnospiracea incertae sedis functioned synergistically with Fibrobacter succinogenes in the fiber digestion [22]. However, Lachnospiracea incertae sedis was poorly characterized phylogenetically and physiologically because of the difficulty of isolation [23]. Therefore, its role in fiber digestion requires further research. The function of the unassigned genus in Firmicutes (phylum) is unknown. We assumed that it might be a novel microorganism involved in fiber digestion. Further work is needed to confirm this speculation.
In the present study, phylum Proteobacteria had significantly lower abundance in AF (1.19%) than that in CF (7.24%). It was reported that Proteobacteria had the highest abundance in the rumen of newborns (1 to 3 d) fed with colostrum and 2-month-old calves fed with milk-supplemented ration, and its abundance sharply decreased in the rumen of the 6-month-old and 2-yr-old cows [2]. It is obvious that the fiber content was higher while the protein content was lower in the ration of the 6-month- and 2-yr-old cows compared with the ration of the calves. Similarly, the protein content was lower while neutral detergent fiber and acid detergent fiber contents were substantially higher in AF than that in CF (Supplementary Table S1). Therefore, it might be a reasonable speculation that the abundance of Proteobacteria would decrease with the increase of fiber content and was positively correlated with the protein content in the ration. Analysis at the genus level seemed to support this speculation, as the major genus within phylum Proteobacteria whose abundance was significantly higher in CF than that in AF was Pseudomonas. It was reported that many strains of Pseudomonas could produce keratinase, which possessed strong protein-degrading function [24].
At genus level, Prevotella was shared by all samples and had the highest abundance both in CF and AF in our study (Table 4), which was in accordance with previous studies in other ruminants [3,9]. Analyzing the previous experiments, we found that the abundance of Prevotella did not always demonstrate regulated changes with the alteration of ration. For example, Huo et al [25] found that the abundance of Prevotella in the rumen from hay-fed goats was higher than concentrate-fed animals. However, Bekele et al [3] found that although the abundance of Prevotella in the rumen of sheep did not differ significantly in the various ration groups, it showed an increasing trend in the concentrate-fed group compared to the roughage group, which was consistent with our results. In our research, although the abundance of Prevotella was numerically higher in the CF, the difference was not significant between the two groups. There are many species among Prevotella which are involved in the degradation of hemicelluloses, pectin, starch, protein, respectively [26]. Due to the various Prevotella species and their different substrate preference, the changes in ration might not always induce regular variation in the abundance of Prevotella.
The study showed that the abundance of Anaerovibrio was significantly higher in CF than that in AF (Table 3). This was probably due to the high-fat content in ration of CF (Supplementary Table S1). Anaerovibrio is a well-known rumen lipolytic bacterium. In the study of microorganisms in rumen of steers, it was found that the relative content of Anaerovibrio in the rumen liquid tended to increase when the oil content in red clover ration increased [27]. In the present study, the Eubacterium content was significantly higher in AF than that in CF, which might be due to the high-fiber content in AF. Eubacterium was mainly involved in the hemicelluloses digestion in the rumen of ruminant and most of the bacteria in this genus could digest xylan [28]. Moreover, it was reported that the abundance of Eubacterium in the rumen of grass-fed cattle was significantly higher than that in the concentrate-fed group [29], which was in accordance with our results.
CONCLUSION
The diet had a significant effect on ruminal microbiota in goats. At genus level, the abundance of fiber-, protein-, and fat-digesting bacteria tended to increase with the rise of their corresponding substrate content in the ration. However, the abundance of some genera shared by all samples such as Prevotella remained stable regardless of the changes in ration. They might be the essential microorganisms that maintained normal digestive function of the rumen. Moreover, many unassigned species whose function in the rumen still needs further investigation were identified in our studies.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print







