




INTRODUCTION
Composting is an effective approach to coordinate the development of pig farms with the surrounding environment (Guo et al., 2007). Composting is a biological aerobic decomposition process that can convert biodegradable organic matter into a humus-like product (Zhu et al., 2004). The key player in organic matter degradation is the natural occurring microbial community inside the compost pile (Maeda et al., 2010; Drury et al., 2013; Lu et al., 2014). A typical composting process is driven by dynamic changes in microbial communities under suitable physico-chemical conditions, and each period is characterized by the growth and development of different microbial communities (Cho et al., 2008).
With the current development of technology, culture-independent molecular methods have been utilized to clarify the molecular phylogenetic diversity and succession of microbial communities present within the composting process. Polymerase chain reaction (PCR) denaturing gradient gel electrophoresis combined with restriction fragment polymorphism (-RFLP) was used to analyze the diversity of the Bacillus community and its temporal-spatial distribution during swine manure composting (Yi et al., 2012a), and the temporal and spatial distributions of Bacillus and Clostridium histolyticum were determined by fluorescent in situ hybridization in their studies (Yi et al., 2012b). The structure of bacterial and fungal communities as well as how these communities change through time, was investigated using high-throughput sequencing (Neher et al., 2013). Their results revealed insights and knowledge on compost microbial communities.
Most of the previous research on the diversity and succession of microbial communities during composting only focused on the hot stage and entire composting period (Beffa et al., 1996; Blanc et al., 1999; Dees and Ghiorse, 2001; Neher et al., 2013). Few studies have been conducted to reveal the diversity and spatial distribution of bacterial communities in the cooling stage. There are changes in temperature, the ration of carbon-nitrogen, potential of hydrogen (pH), total organic carbon, organic matter, total kjeldahl nitrogen, water soluble carbohydrate that occur during the various stages of the composting process. Consequently, the bacterial community is different in the special cooling stage during the swine manure composting process. Significant discoveries regarding this stage of the process could promote our understanding of the bacterial community. The results may help us reveal the panorama of the bacterial community in the complex composting process, and how to control it.
This study aimed to investigate the diversity and spatial distribution of a bacterial community in the cooling stage during the swine manure composting process using a culture-independent method (i.e. RFLP) and subsequent sub-cloning and sequencing, and identify the dominant bacteria within each layer of a passively aerated compost pile.
MATERIALS AND METHODS
Composting and sampling
A 1.5 m×1.2 m×1.2 m (length×width×height) composting bin was designed and built. The bin was covered by a shed to shield it from sun and rain. For the reactor, ~500 kg of swine manure and 25 kg of rice straw were mixed according to an initial carbon nitrogen ratio of 20. The initial moisture content of the mixture was adjusted to about 60%.
Temperatures at different locations of the pile, including substrate (20 cm from bottom), middle-level, and surface (20 cm to surface) temperatures, and environmental temperature were monitored by Ttime system (Zhu et al., 2003). The temperature data were refurbished per 10 s, and saved automatically per hour.
About 1kg of fresh composting sample was collected at different locations, namely, substrate (20 cm from bottom), middle-level, and superstratum (20 cm to surface), when the middle-level temperature reached 40°C in the cooling stage. The exact temperatures of the superstratum and substrate layers were 39.7°C and 33.7°C, respectively. The collected samples were immediately placed on ice for transportation to the laboratory and then stored at –20°C.
Extraction and purification of total DNA
A modified DNA extraction protocol was described by the researchers (Holben, 1994; Dees and Ghiorse, 2001). A sample (10 g) of the frozen compost was added to 90 mL of 0.1% NaPP, and shaken for 30 min at room temperature on an orbital shaker at ~150 rpm. The mixture was transferred to a sterile Waring blender jar, and homogenized using three 30 s bursts with a 30 s pause between blending steps. Straw and debris were left in the blender jar, whereas the remainder of the homogenized slurry was transferred to 35 mL centrifuge vials and centrifuged for 20 min in a Beckman Model Avanti J-20 (San Francisco, CA, USA) using a JA-17 rotor at 12,000×g. The blender cup containing straw and debris was rinsed with an additional 35 mL of NaPP. The resulting slurry was transferred to a fresh 35 mL centrifuge vial and centrifuged for 20 min at 12,000×g. The pelleted material from the reinstate was combined with the pellet from the first step. Straw and debris were intentionally left in the slurry to include organisms that might remain tightly attached to compost particle surfaces. The combined pellets from the centrifugation steps were frozen at –70°C in a refrigerator (ULT1386-3-V35, Asheville, NC, USA).
For cell lysis and DNA purification, 50 or 100 mg of the above frozen material was added to a 2.2 mL centrifugal vial containing ~1.2 g of 0.1 mm diameter silica/zirconia beads. The vials received 300 μL each of the following components: sodium phosphate buffer(100 mM NaH2PO4, pH 8.0), lysis buffer (100 mM NaCl, 500 mM Tris, pH 8.0, 10% sodium dodecyl sulfate), and chloroform-isoamyl alcohol (24:1) mixture. The vials were agitated for 5 min at maximum speed (~2,500 rpm) and then were centrifuged briefly at 3,000 rpm in a microcentrifuge to settle the contents. The aqueous supernatant containing the community DNA was transferred to another sterile 1.5 mL microcentrifuge vial and further purified using a UNIQ-10 DNA Purification Kit (Bio Basic Inc, Shanghai, China), following the manufacturer’s instructions except for the first step. The extracted crude DNA was joined into the column directly, placed for 2 min in room temperature, and following the manufacturer’s instructions. Purified DNA was stored at –20°C for PCR amplification.
Polymerase chain reaction amplification and cloning
Bacterial 16S rRNA genes were amplified by PCR using the universal primer pair 1492r (5′ TAC CTT GTT ACG ACT T 3′) and 27f (5′ AGA GTT TGA TCC TGG CTC AG 3′) (Lane 1991; Dojka et al., 1998). The total sum of the component of the PCR mix exceeded 25 μL: 10 pmol of both primers, 3 μL of genomic DNA, 1.5 μL of 2 mM mixed dNTPs, 2.5 μL of 10×Taq DNA polymerase buffer, 2.5 μL of 25 mM MgCl2, 2.0 U of Taq DNA polymerase (1 U/μL) (Jingmei Biotech, Shenzhen, China), and 15 μL of sterile water. PCR was performed under the following conditions: 30 cycles of 94°C for 40 s, 52°C for 45 s, 72°C for 2 min, extension at 72°C for 10 min, and 4°C to terminate the reaction. The amplified products were electrophoretically separated and visualized in 1.5% agarose gels stained with ethidium bromide. The interesting band was removed from the gel and purified with a UNIQ-10 DNA Purification Kit (Bio Basic Inc, China). Finally, the purified product was ligated into the pMD 18-T Vector (Takara, Dalian, China), and the ligation product was transformed into Escherichia coli DH5α competent cells. White transformant colonies (Sambrook et al., 1989) were selected randomly, and the vector insert size was detected using M13/pUC universal primers P47 and P48.
Restriction fragment length polymorphism screening
Plasmids were extracted from the positive transformant colonies, and subjected to RFLP analysis by separate endonuclease digestions with HhaI (Mentas) and HaeIII (Mentas) following the manufacturer’s instructions. The digested DNA fragments were electrophoresed in 2.5% agarose gels. The gels were photographed after staining with ethidium bromide, and scanning image analyses were performed manually.
DNA sequencing and phylogenetic analysis
One to three representative clones from each unique RFLP type were selected for sequencing. The rDNA inserts were sequenced using M13/pUC universal sequencing primers P47 and P48. All near full-length sequences were tested for possible chimeric structures. Chimeras were detected by the ribosomal database project analysis service Check-Chimera by comparing the phylogenetic positions of near full-length sequences from the clones and during manual inspection of the alignment.
The spelled nearly entire sequences (~1,500 bp), except for the chimeras, were aligned using the Clustalw program (http://www.ebi.ac.uk/clustalw/). The sequences were assigned to individual operational taxonomic units (OTUs) based on a sequence similarity of at least 97%. Each OTU was compared with those available in GenBank by performing BLASTn program (http://www.ncbi.nlm.nih.gov) to determine their approximate phylogenetic affiliation. Phylogenetic trees were constructed based on the Kimura two-parameter model and neighbor-joining algorithm using the PAUP software (version 4.0 b8) (Swofford, 1999).
RESULTS
Polymerase chain reaction amplification and clone library analysis
Total community genomic DNA extracted from the three layers (superstratum, middle-level, and substrate) of composts was subjected to PCR amplification using the primer pair (27f/1492r). The ~1,500 bp PCR product was used to create a 16S rDNA clone library, and three bacterial 16S rRNA gene libraries were established to represent the superstratum, middle-level and substrate composts. To exclude false positives, 150 recombinant clones, called swine manure G sample (SMG), swine manure Q smple (SMQ), and swine manure R sample (SMR), from each sample were randomly selected and screened by restriction endonuclease analysis. From the 150 clones, 78, 63, and 53 unique RFLP types were identified, and 11, 1, and 18 false-positive clones were excluded from the superstratum, middle-level, and substrate samples, respectively. One representative of each of the RFLP types was selected for sequencing, and phylogenetic analysis was conducted on these near full-length 16S rDNA sequences.
Bacterial diversity determination
Under the criterion 97% sequence similarity, 48, 26, and 35 OTUs were found in the superstratum, middle-level, and substrate samples, respectively. Each of OTU represents a phylotype and may be a representative of a bacterial species. The number and distribution of OTUs, as well as bacterial diversity, varied among the three different sampling locations.
Isolates and cloned sequences of the superstratum compost
In this group, 139 clones were sequenced across 1,500 bp. A total of 78 sequences were classified into 48 OTUs with at least 97% similarity. Within these bacterial clones, 107 were represented by 16 OTUs with at least two clones. The other 32 OTUs were detected with only one clone. The number of clone(s) of each OTU was marked in the bracket after SMG in the phylogenetic tree (Figure 1).
Eleven sequences classified into six OTUs belonged to the Bacillus group. Eight sequences classified into three OTUs belonged to the Paenibacillus group. Seven sequences classified into three OTUs representing the largest group, including 35 clones, belonged to Thermobifida fusca. Five sequences classified into two OTUs belonged to the Sphingomonadaceae group. Five sequences classified into two OTUs belonged to Pseudomonas fluorescens. Four sequences classified into two OTUs belonged to the Bordetella group. Four sequences classified into three OTUs belonged to the Clostridium group. Three sequences classified into one OTU belonged to Pusillimonas noertemannii. Three sequences classified into one OTU had no similarity to the known species. Two sequences classified into two OTUs belonged to the Alcaligenes group. Two sequences classified into two OTUs belonged to Providencia stuartii. Two sequences classified into two OTUs belonged to Moorella glycerini. Two sequences classified into one OTU belonged to Saccharomonospora viridis. The other two sequences classified into one OTU belonged to Planifilum fimeticola. The remaining 18 sequences classified into 18 OTUs were minor groups in the clones analyzed, and one of those clones was not closely related to any known species.
Isolates and cloned sequences of the middle-level compost
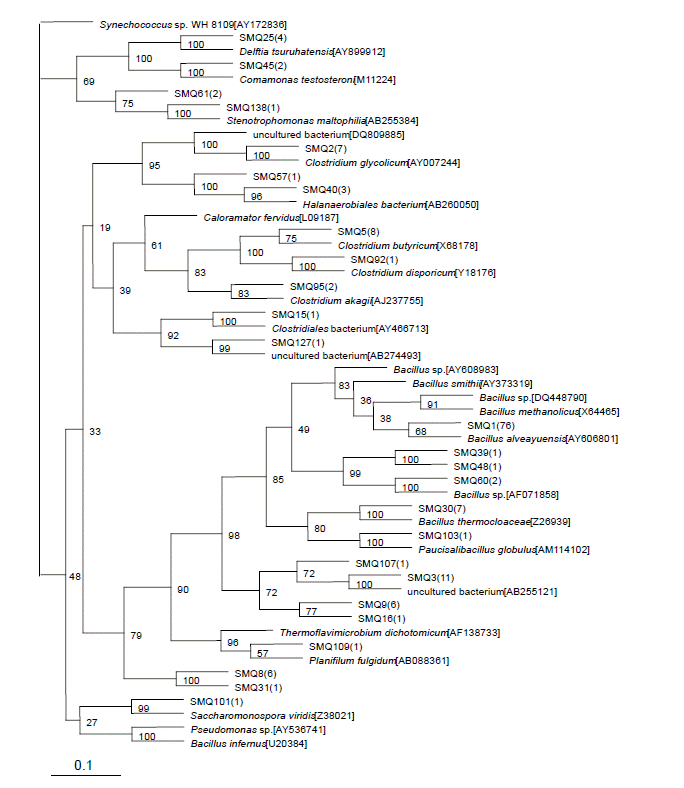
In this group, 149 clones were sequenced across 1,500 bp. A total of 63 sequences were classified into 26 OTUs with at least 97% similarity. Within these bacterial clones, 136 were represented by 13 OTUs, including at least two clones, whereas the other 13 OTUs were detected with only one clone. The number of clone(s) of each OTU was indicated in the bracket after SMQ in the phylogenetic tree (Figure 2).
Twenty-eight sequences classified into eight OTUs representing the largest group, including 95 clones, belonged to the Bacillus group. Fifteen sequences classified into six OTUs belonged to the Clostridium disporicum group and relatives. Four sequences classified into one OTU belonged to Caloramator fervidus. Four sequences classified into one OTU belonged to Delftia tsuruhatensis. Three sequences classified into one OTU had no similarity to known species. The remaining nine sequences classified into nine OTUs were minor groups in the clones analyzed, and one of those clones was not closely related to any known species.
Isolates and cloned sequences of the substrate compost
In this group, 132 clones were sequenced across 1,500 bp. A total of 53 sequences were classified into 35 OTUs with at least 97% similarity. Within these bacterial clones, 111 were represented by 14 OTUs, including at least two clones, whereas the other 21 OTUs were detected with only one clone. The number of clone(s) of each OTU was indicated in the bracket after SMR in the phylogenetic tree (Figure 3).
Twenty-six sequences classified into 14 OTUs representing the largest group, including 89 clones, belonged to the Clostridium group. Four sequences classified into four OTUs belonged to the Bacillus group and relatives. Four sequences classified into one OTU had no similarity to the known species. Two sequences classified into two OUTs belonged to the Lactobacillus group. Two sequences classified into one OTU belonged to Holdemania filiformis. The remaining 15 sequences classified into 13 OTUs were minor groups in the clones analyzed, and 15 of those clones were not closely related to any known species.
The bacterial communities of Clostridium and Bacillus were mainly detected in the three different layers in the cooling stage, and the relative abundance differed among the three composting samples. Based on 16S rDNA sequence analysis, the diversity of Bacillus in the middle-level sample at the cooling stage was the highest, but that of Clostridium sp. was remarkable in the substrate sample. These results strongly support those of a previous study (Yi et al., 2012b), in which Bacillus and Clostridium were found to be predominant in swine manure composting. In addition, the specific bacterial community was detected in the three different location samples. Our results suggest that the phenomenon of spatial distribution in a microbial community caused by temperature was common and desirable during composting.
DISCUSSION
In this study, 420 clones were subjected to PCR-RFLP and sequencing analysis. A total of 170 clones (40.48%) showed high levels of sequence relatedness (≥97%) with 14 known species, whereas 216 clones (51.43%) had a sequence similarity of less than 97%. The remaining 34 clones (8.10%) showed high levels of sequence relatedness (≥97%) with the sequences in the database not assigned to any genera, indicating that a portion of bacterial species associated with composting remains to be identified. The 78 sequences derived from the superstratum compost were classified into 48 OTUs with at least 97% similarity. Among these 78 sequences, 74 belonged to Firmicutes (44.87%), Bacteroidetes (5.13%), Actinobacteria (11.54%), and Proteobacteria (33.33%), and the remaining four sequences (5.13%) were not assigned to any genera. In the middle-level group, 63 sequences were classified into 26 OTUs. Among these 63 sequences, 59 belonged to Firmicutes (80.95%), Proteobacteria (11.11%), and Actinobacteria (1.59%), and the remaining four sequences (6.35%) were not assigned to any genera. However, the 53 sequences derived from the substrate compost were classified into 35 OTUs with at least 97% similarity. Among these 53 sequences, 41 were classified into Firmicutes (62.26%), Bacteroidetes (1.89%), Proteobacteria (9.43%), and Tenericutes (3.77%), and the remaining 12 sequences (22.64%) were not assigned to any genera.
This study provided an overall picture of the diversity and spatial distribution of bacterial communities, and revealed the domain bacteria belonging to the Bacillus and Clostridium populations in three different locations of the cooling stage during the swine manure composting process. The bacterial communities in the superstratum layer displayed higher relative abundance and diversity than the bacterial communities in the middle-level and substrate layers (Figures 1, 2, and 3). The number of OTUs in the superstratum layer was the highest, whereas that in the substrate layer was he lowest. Regarding the spatial distribution of bacterial communities, bacteria related to the genus Bacillus were the most common in the middle-level layer. The most abundant OTUs in the substrate layer were related to the genus Clostridium. However, T. fusca was the most commonly detectable bacterium in the superstratum layer. Thus, the three clone libraries differed significantly from one another. The diversity and spatial distribution were indeed affected by physic chemical factors, such as temperature, pH, O2, and CO2 concentrations.
Ten 16S rDNA sequences were not closely related to any known species. Although these bacteria exhibited a low frequency in the constructed gene libraries, the following interesting question was raised: “What role do these clusters play in the composting process?” More studies are necessary to clarify the actual importance of these bacteria in the initial stage of swine manure composting. The results provided a deeper insight into the diversity and spatial distribution of bacterial communities at different locations in cooling stage during the composting process. Molecular biology techniques based on 16S rDNA sequence analysis are a useful for studying the diversity and spatial distribution of microbial communities in swine manure composting. With the development of science and technology, other new molecular techniques can be utilized to describe the environmental microbial community. An increasing number of microorganisms can be found and studied, but only a small fraction (0.01% to 10%) of microorganisms in natural environments have been identified (Ward et al., 1992; Amann et al., 1995; Torsvik et al., 1996). In conclusion, the results of our research could help us understand complex processes and further enhance the efficiency of swine manure composting.
Compared with the other two stages, the diversity and dominant microflora are different. In the initial stage, the micro-organism species in the middle-level compost were more diverse than the other two layers, and the number of clones related to the genera Clostridium is dominant in middle-level and substrate samples, but Shigella sp. in the superstratum sample (Guo et al., 2007). In the high-temperature stage, Bacillus sp. (45 clones, 33%) and Clostridium sp. (36 clones, 27%) were the main microorganisms in the superstratum sample, 74 clones (58%) were affiliated with the Clostridium sp. in the middle-level sample; 52 clones (40%) and 29 clones (23%) were affiliated with the Clostridium sp. and Bacillus sp. in the substrate sample, respectively (Guo et al., 2012). It’s worth mentioning that Clostridium constantly keep predominate in the whole composting process.
The data indicated that the microbial diversity and community in the samples were different for each sampling site, and different locations of the same pile often contained distinct and different microbial communities. The results of these studies revealed the succession of microbial community during composting process, and the microorganism community diversity among the different location samples in the different stages provided the biology evidence of the phenomenon “gradient effect”.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







