




INTRODUCTION
In pigs, nipples are essential for nursing babies during lactation. For a given number of piglets, the sows with more functional teats produce more weaned pigs as compared with those having fewer functional teats. Thereby, teat number is an economically important trait that significantly affects reproduction efficiency in pigs.
Unlike most reproductive traits, teat number is an inborn trait that is less affected by environmental factors [1]. Different pig breeds have variable teat, numbers, ranging from 8 to 25. The genetic architecture of teat number is very complicated. To date, 120 quantitative trait loci (QTLs) have been identified for teat number which span almost all pig chromosomes (http://www.animalgenome.org/cgi-bin/QTLdb/SS/index). Recently, genome-wide association studies (GWAS) have been conducted to map loci affecting pig teat number and have identified 11 interesting candidate genes for this trait [2–4]. However, the responsible genes and causative mutations for the identified loci remain to be characterized. Moreover, QTLs for pig teat number in a broad context of diverse breeds warrant further investigations by using the advanced GWAS tool.
In this study, we conducted GWAS to map loci affecting teat number on three pig populations, including a White Duroc×Erhualian F2 resource population (n = 1,743), a Chinese Erhualian population (n = 320) and a Chinese Sutai population (n = 383). The results improve our understanding of the genetic architecture of pig teat number, and provide novel molecular markers for the genetic improvement of this reproductive trait in the pig industry.
MATERIALS AND METHODS
Animals and phenotypic recording
This study was conducted strictly in compliance with guidelines of experimental animals established by the Ministry of Agriculture of China, and was approved by the ethics committee of Jiangxi Agricultural University (China).
Experiment animals were from three pig populations, including the White Duroc×Erhualian F2 resource population, Chinese Erhualian and Sutai populations. The F2 resource population was established by intercrossing 2 White Duroc boars (PIC 1075) and 17 Erhualian sows as described previously [5]. In brief, a total of 1,912 F2 pigs were generated by 9 F1 boars and 59 F1 sows in 6 batches, avoiding full-sub mating. We recorded 1,743 F2 pigs for teat number. Erhualian is a highly prolific Chinese indigenous pig breed characterized by its favorable maternity and high litter size. We brought 320 unrelated (no common ancestors within 3 generations) Erhualian pigs from an Erhualian conservation farm in Jiangsu province. Sutai is a Chinese cultivated pig breed that was originally derived from a cross between Chinese Taihu and Duroc pigs and have experienced artificial selection for pork production and fertility for more than 18 generations. A total of 383 Sutai pigs from 5 sires and 60 dams were used in this study. All animals from the three populations were raised indoor in the experimental farm of Jiangxi Agricultural University (China), and were recorded for teat number by simply counting right and left teat number at the weaning period.
Genotyping and quality control
A standard phenol/chloroform approach was used to extract DNA from ear tissues. DNA quantity and quality were determined by a Nanodrop-1000 spectrophotometry (Thermo Fisher, Waltham, MA, USA). All DNA samples were diluted to a final concentration of 50 ng/mL. In our previous study, 1,899 F2 pigs were genotyped for 183 microsatellite markers [6]. Here, 928 F2 pigs and their F1 and F0 ancestors, 320 Erhualian and 383 Sutai pigs were genotyped for 62193 single nucleotide polymorphisms (SNPs) on the Illumina Porcine 60K Beadchip [7]. The 60K SNP data of the 928 F2 pigs and their F0 and F1 ancestors (reference animals) were further employed to impute the 60K SNP genotypes of 815 F2 pigs (target animals) that were not genotyped by 60K SNP chip by using the following approach [8]. Firstly, haplotypes of the reference and target animals were partially reconstructed based on linkage and Mendelian segregation rules with the 183 microsatellite data using DualPHASE [9]. Secondly, haplotypes of the reference individuals were fully reconstructed with microsatellite data and 60K SNPs data using DAGPHASE, which was iteratively called by Beagle [9]. Lastly, missing 60K SNP genotypes of the target pigs were filled via CHROMIBD, in which the linkage and a Markov model were utilized to estimate identity-by-descent (IBD) probabilities between target and parent chromosomes from the genotyped ancestors [10].
The quality control for real and imputed genotypes SNPs were carried out by the Plink 1.07 software [11]. Animals with SNP call rates ≤0.9, SNPs with call rates ≤0.9, minor allele frequencies (MAF) ≤0.05, p-values ≤10–6 for the Hardy-Weinberg disequilibrium test and Mendelian error rate ≥ 0.1 were removed for further statistical analysis. A final set of 31,557, 27,719 and 43,998 SNPs were remained for GWAS in the F2, Erhualian and Sutai populations, respectively.
Statistical analysis
A principal component analysis (PCA) was conducted to test population structures of the three populations. We generated the PCA plots by using the –mds-plot and –cluster options in Plink and visualized it via the R program. The GWAS was performed with a linear mixed model in GenABEL v1.8, a library of R program [12]. The GWAS model is shown as below:
Where y represents the vector of phenotypes of all tested pigs; β is the fixed effects, including all grand mean and sex; α is the regression coefficient of substituting allele; u is the vector of polygenic effect; e is the vector of residual errors. X and Z are incidence matrices for β and u, respectively. S is the number of substituting alleles.
Q-values were computed by the q-value package of R language and were then used to estimate the false discovery rate (FDR) threshold for the significant p-value of 0.05 [13, 14]. Confidence regions were determined by log of odds ratio (LOD) dropoff 2 from the peak SNPs. A meta-analysis of GWAS was performed to combine evidences of true associations from the three populations with appropriate weight via METAL [15]. The meta-analysis was implemented with an inverse variance based strategy that weighted on the β-coefficients [15]. A common set of 16,396 SNPs across these populations were used in the meta-analysis.
Annotation of candidate genes
The porcine genome assembly 10.2 (http://www.animalgenome.org/repository/pig/Genome_build_10.2_mapping/) and National Center for Biotechnology Information (NCBI) Genome (http://www.ncbi.nlm.nih.gov/genome/?term=pig) were retrieved to characterize candidate genes in targeted regions. Function and signal pathway of annotated candidate genes were found via Ensembl Biomart (http://www.biomart.org/), GeneCards (http://www.genecards.org/), NCBI Gene (http://www.ncbi.nlm.nih.gov/gene/) and Cellsignal (http://www.cellsignal.com/).
RESULTS
Phenotypic data and population stratification
Descriptive statistics of teat number in the three tested populations are shown in Table 1. The total teat number of Erhualian pigs ranged from 14 to 25, with an average number of 20.38. In contrast, Sutai pigs had the lowest average number of teats (15.20). The PCA analysis indicated that no population stratification existed between the 928 F2 pigs with real 60K SNP genotypes and the 815 F2 pigs with the imputed data in the White Duroc×Erhualian F2 population, suggesting the reliability of the imputation method. Nevertheless, the F2, Erhualian and Sutai populations displayed apparently different population structures, reflecting their divergent genetic background (Supplementary Figure 1).
GWAS results
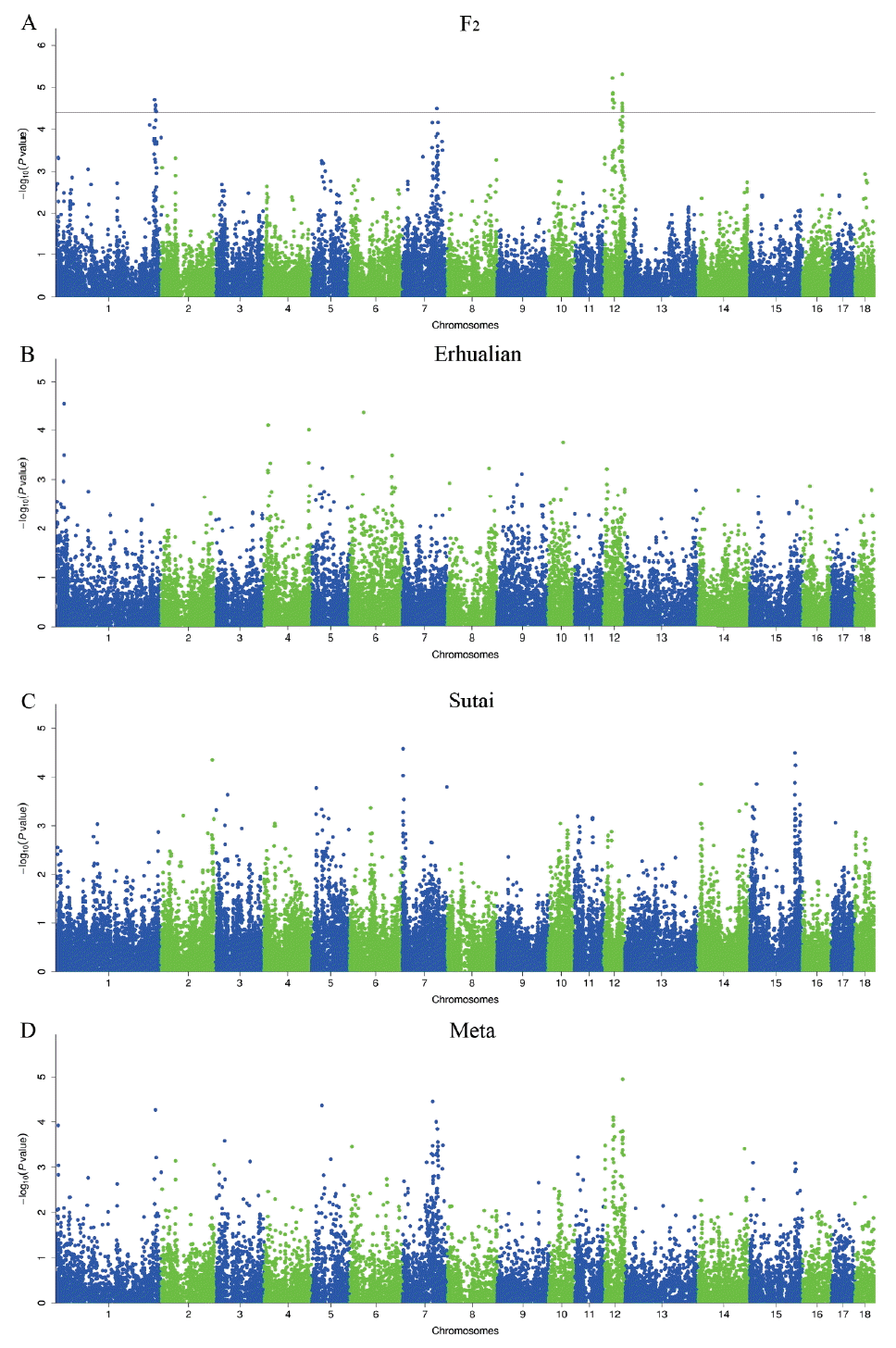
The average inflation factors (λ) of GWAS ranged from 0.961 to 1.028, indicating that all population structures were properly adjusted (Supplementary Figure 2). In total, we detected 24 SNPs associated with porcine teat number in the F2 resource population, including the loci around 295.04 Mb (293.78 to 301.56 Mb) on Sus Scrofa chromosomes (SSC) 1, around 103.91 Mb (89.15 to 106.91 Mb) on SSC7, around 26.02 Mb (26.02 to 31.88 Mb) and 55.88 Mb (45.75 to 56.79 Mb) on SSC12 (Table 2 and Figure 1). No significant association was found in the Erhualian and Sutai populations (Figure 1). Besides the above-mentioned significant loci, we noted that the SNPs around 25.30 Mb on SSC1 and 42.95 Mb on SSC6 in Erhualian pigs, and those around 3.51 Mb on SSC7 and 135.71 Mb on SSC15 in Sutai pigs showed potential associations with p-values close to the significant threshold (Figure 1). The loci on SSC1 and SSC6 in Erhualian pigs has also been reported to be associated with teat number in a Meishan×Gottingen cross population and a Large White×Meishan F2 population [16, 17]. In the meta-analysis of GWAS, no SNP showed significant association with teat number (Table 2 and Figure 1). Ten SNPs reached the 0.20 q-value threshold, suggesting a minimum of ~19% FDR for these SNPs if judged as significant ones.
DISCUSSION
Genotype imputation and meta-analysis for GWAS
More currently, genotype imputation and meta-analysis have been explored in GWAS of genetic diseases or phenotypic traits [15,18,19]. Genotype imputation is a reliable tool to equate different sets of markers from multiple platforms [18]. With the development and improvement of multiple imputation algorithms, missing genotypes can be imputed with high accuracy rates [20]. In the White Duroc×Erhualian F2 resource population, we have estimated an average of 91.44% of genotype concordance, 95.36% of allelic concordance and 0.85 of r2 correlation between real and imputed data using a cross-validation analysis [21]. By applying an imputation based GWAS, we have successfully identified a list of significant loci for traits related to male reproduction in this F2 population [21]. Meta-analysis of GWAS provides a robust tool to identify true associations by pooling data from different studies or populations [15]. This approach enables researchers to efficiently account for population substructure, studies-specific covariates, and individual relatedness [15].
In this study, we implemented the imputation method to deduce the 60K SNP genotypes of 815 F2 pigs using the genotyped 928 F2 pigs and their F0/F1 ancestor as the reference panel. The markers on sex chromosomes were not imputed for a lack of an appropriate algorithm [22]. This imputation approach allowed us to increase the sample size up to 1,743 animals in the F2 resource population. In the meta-analysis of GWAS, we did not identify any SNP that surpassed the significant threshold. One explanation could be that different populations have distinct causative mutations for teat number; thus, the meta-analysis cannot increase but decreases the detection power. Another possible reason is that the qualified SNPs in the meta-analysis may be in different linkage disequilibrium phases with causative mutations in different populations, leading to inconsistent allelic directions and consequently reducing the detection power.
Comparison of GWAS loci with previously identified QTLs
Teat number is a complex reproductive trait. A total of 120 QTLs have been identified for this trait on almost all pig chromosomes except for SSC13, suggesting that teat number is controlled by multiple genes with generally small effects. We have previously identified 11 genome-wide significant QTLs for teat number by a genome scan on the White Duroc×Erhualian F2 resource population [6]. These loci were distributed on SSC1, 3, 4, 5, 6, 7, 8, and 12, which were consistent with previous reports [1, 23–29]. Of the 11 significant loci, the loci on SSC1, SSC7 and SSC12 were replicated in this study (Table 2 and Figure 1). We note that the loci around 102 to 105 Mb on SSC7 and around 52 to 56 Mb on SSC12 were also consistently detected in a Landrace-based line and a Large White line [2,3]. However, those QTLs on SSC3, SSC4, SSC5, SSC6, and SSC8 that we detected in our previous study were not evidenced by the GWAS in this study. This may be due to the following points. First, the GWAS thresholds including Bonferroni-corrected and FDR-defined thresholds are more stringent than the QTL threshold. Second, the traditional QTL mapping conducts stepwise conditional analysis, i.e. the effect of QTL detected in a previous round was treated as a fixed effect in the statistical model when searching for QTL in the next round. This approach has smaller error residuals and consequently has a stronger detection power than the linear mixed GWAS model [30]. Third, only additive effect were included in our GWAS model while both additive and dominant effects were considered in the QTL model. For example, the QTL on SSC6 has been identified as a significant locus for teat number with both additive and dominant effects in the F2 population [6]. Finally, the underlying assumptions of QTL and GWAS were not same. In the QTL mapping study, the QTL was assumed to be fixed in alternative alleles in the two found breeds, respectively, and the markers of three generations (founders, F1 and F2) were used to track the QTL genotype in the F2 animals; while the SNP genotypes were considered as causative SNP genotypes in GWAS. Although we detected few significant loci in the GWAS, we obtained important clues for the most likely locations of these loci by using high-density SNPs across the genome as compared to sparse microsatellite makers used in the QTL mapping (see below). We note that two regions are associated with teat number on SSC12 where only one QTL was identified in our previous study [6]. This could be due to the fact that only 7 microsatellite markers with an average interval distance of 9.44 Mb on this chromosome were used for mapping QTLs, which profoundly limited the mapping resolution of the traditional QTL analysis.
Comparison of significant loci across the three tested populations
We did not identify any significant locus that was shared by the three tested populations or two of these populations (Figure 1). Although the F2 population was originally derived from a cross between White Duroc and Erhualian breeds, the four significant loci detected in the F2 population did not appear in the purebred Erhualian population. These loci are likely caused by variants that are alternatively fixed (or nearly fixed) in Duroc and Erhualian pigs. Sutai pigs are descendants of a hybrid between Duroc and Taihua (Erhualian is a subgroup of Taihu pigs), mimicking the genetic makeup of the F2 population. No common significant locus was evidenced between the two populations, either. These observations further reflected the complex genetic architecture of pig teat number. It should be mentioned that MAF of many chip SNPs varied considerably in the three populations. For example, all nine significant SNPs on chromosome 1 were fixed in the Erhualian population (data not shown). Hence, we cannot rule out the possibility of false positive or negative results that are caused by variable MAF in this study.
Plausible candidate genes at the significant loci
For significant loci that we identified, we searched plausible candidate genes within the confidence region of each locus. Combining our results with the earlier publications, we propose lysine demethylase 6B (KDM6B) and vertnin (VRTN) as two interesting candidate genes for the loci on SSC12 and SSC7. No annotated gene was found in the other QTL regions that are functionally related to the development of teat. KDM6B is 0.42 Mb away from the top SNP (ALGA0121951) at the genome-wide significant locus on SSC12. This gene plays a critical role in guiding the expression of T-box gene family in embryonic stem cell differentiation [31]. T-box3, a member of the T-box gene family, has been reported to influence the development of mammary gland placodes via interacting with Wnt family member 10 (Wnt10), lymphoid enhancer binding factor 1 (lef-1), and fibroblast growth factor 10 (Fgf10) [32–34]. Moreover, T-box3 variants have been implicated in the Ulnar-Mammary Syndrome in humans, a genetic disease characterized by nipple and breast hypoplasia or aplasia [35,36]. Given that KDM6B is located in the confidence interval of the genome-wide significant locus and has a potential role in the development of nipples, we highlight it as an interesting candidate gene for pig teat number that warrant further investigations.
VRTN is 0.71 Mb away from the top SNP (H3GA0022664) at the genome-wide significant locus on SSC7 in the White Duroc×Erhualian F2 resource population (Figure 1). This gene is known to play an essential role in cell differentiation of embryos in different species and has a major effect on the development of thoracic vertebrae (ribs) in pigs [37,38]. Vertebral number is positively correlated with body size in pigs. Hogs with more vertebrae tend to have longer body length and better meat production capacity. Selective breeding for enlarging body size to increase meat production has been carried out for decades in Western commercial breeds, resulting in increased vertebral number and presumably more teats [3]. Therefore, VRTN has been selected as a promising candidate gene for the number of nipples by several other research groups [2,3]. Here, we observed that teat number is significantly correlated with vertebral number in the F2 population (r = 0.21, p-value = 1.51×10–10). When conditional on the effect of a candidate causative mutation of VRTN (g.20311_20312ins29) [37, 38] in the F2 population, the GWAS signal at the SSC7 locus vanished in this population (data not shown). Hence, we assume that causative mutation(s) in the VRTN gene may have pleiotropic effects on the teat number and vertebral number. Recently, we shown that the VRTN mutation is significantly associated with teat number in a large sample of Western Duroc and Landrace pigs [39]. This finding has an immediate transition into the pig breeding schemes, providing a novel tool to improve both teat number and vertebral number by selection of the desirable allele at the VRTN locus. We note that no significant association signal was detected in the VRTN region in the Erhualian and Sutai populations. One reasonable explanation is that the causative variant(s) in the VRTN gene is fixed or very rare in the two populations.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print







