




INTRODUCTION
Postmortem glycolysis is one of the most important factors determining the quality of meat. Abnormal glycolysis in postmortem muscle induces meat defects, like PSE (pale, soft, exudative), DFD (dark, firm, dry) and acid meat. Among these problems, PSE syndrome is the most serious one to meat industry. PSE meat has a high drip loss, a low cooking yield, and a tough texture after cooking. Surveys indicated that of pork produced in the USA, 15.5% was PSE meat in 2002 and 3.34% was typical PSE meat in 2005 [1]. Due to the undesirable appearance and limited function in processing, PSE meat is not preferred by consumers and estimated to cause an annual loss of millions of dollars in the USA [1].
Since first being described in 1953 [2], PSE meat has been recognized for more than half a century, but remains a challenge to the industry. Early studies established that the development of PSE pork is largely due to an increased rate of glycolysis early postmortem, as indicated by elevated muscle temperature and rapid pH decline [3]. Subsequently, numerous studies revealed that pre-slaughter stress and genetics are the two main factors contributing to PSE meat [4ŌĆō6]. Thus, for the prevention of PSE meat, it is critical to control the rate of glycolysis in early postmortem muscle, but questions remain regarding the biochemistry of postmortem glycolysis and the molecular mechanisms through which the normal energy metabolism of postmortem muscle is maintained or disrupted by pre-slaughter practices and genetic factors that ultimately lead to altered meat quality development.
Previous studies show that AMP-activated protein kinase (AMPK) plays an important regulatory role in postmortem glycolysis [7ŌĆō9], but the downstream mediators that coordinate AMPK activity and glycolytic rate in postmortem muscle are not well documented. Protein acetylation is a common posttranslational modification which may rank second to protein phosphorylation [10]. Since it was first discovered in nuclear histones [11], the enzymes that catalyze protein acetylation and deacetylation are subsequently referred to as histone acetyltransferase (HAT) and deacetylase (HDAC). Since then, histone acetylation and its regulation of gene expression have been well recognized and received intensive study. Recently, it has been reported that protein acetylation and deacetylation plays an important role in cellular metabolism from bacteria to humans. Most enzymes involved in intermediate metabolism have been shown to be acetylated, which include nearly all enzymes involved in glycolysis, glycogen metabolism, gluconeogenesis, the tricarboxylic acid cycle, fatty acid oxidation, the urea cycle and some enzymes involved in oxidative phosphorylation and amino acid metabolism [12,13]. In addition, several studies have reported that AMPK regulates gene expression, mitochondrial biogenesis and energy metabolism through its phosphorylation of HDAC or by regulating deacetylase activity of Sirt1 [14ŌĆō16]. Thus, we hypothesize that protein acetylation plays a role in the regulation of glycolytic enzyme activity, and AMPK regulates the reversible acetylation of glycolytic enzymes through its action on HAT and/or HDAC, which in turn regulates the rate of postmortem glycolysis. The objective of this study was to investigate the influence of AMPK activation on protein acetylation in relationship to glycolysis in postmortem muscle. Since the biochemistry of glycolysis is the same for mice, pigs and poultry and to well control enzyme activities by chemicals, mice were used in this study.
MATERIALS AND METHODS
Animal treatments
All procedures of animal handling were approved by the Chinese Academy of Agricultural Sciences Animal Care and Use Committee. A total of 32 three-month-old C57BL/6J mice (male) were randomly assigned to four groups: i) Control, mice were injected with vehicle before euthanized; ii) AICAR, mice were intraperitoneally injected with 50 ╬╝g/g AICAR (Sigma, St. Louis, MO, USA), a specific activator of AMPK, and waited for 15 min to let the AICAR go into muscle before euthanized; iii) A+HATi, mice were injected with 50 ╬╝g/g AICAR and 185 ╬╝g/g HAT inhibitor II (Sigma, USA), and waited for 15 min before euthanized; iv) A+HDACi, mice were injected with 50 ╬╝g/g AICAR, 8.0 ╬╝g/g Trichostatin A (TSA, an inhibitor of HDAC I and II) and 250 ╬╝g/g Nicotinamide (NAM, an inhibitor of the Sirt family deacetylases) (Sigma, USA), and waited for 15 min before euthanized. Animals were anaesthetized by CO2 and killed by cervical dislocation. Within 4 min, the pelt was removed and ~0.3 g each of the upper-right and lower-left Longissimus dorsi (LD) muscle was removed and combined. Part of this muscle (~0.1 g) was used for pH measurement and the rest of muscle was snap-frozen in liquid nitrogen for the analysis of enzyme activity, glycolytic potential and western blotting. Carcasses were eviscerated and placed at 4┬░C. Subsequent samples were taken in a similar fashion from the middle parts of both sides of the muscle at 45 min and 24 h postmortem. Muscle samples were trimmed free of fat and connective tissue and then snap-frozen in liquid nitrogen and stored in ŌłÆ80┬░C for subsequent analyses.
pH measurement
Once removed, the LD muscle (~0.1 g) was homogenized in 0.9 mL of 5 mM iodoacetate solution immediately. The pH of the homogenate was measured directly with a pH meter.
Lactate analysis
Lactate in muscle was determined as previously described [17]. Briefly, 0.05 g muscle was homogenized in 450 ╬╝L of 0.9 N HClO4. The homogenates were centrifuged at 13,000├Śg, 4┬░C for 5 min. The supernatants were removed and neutralized with 2 M KOH and centrifuged again to precipitate potassium perchlorate. The extracts were used for lactate measurement using a commercial kit (Sigma, USA).
Glycolytic potential
Total concentrations of glycogen, glucose, and glucose-6-phosphate in muscle were determined using commercially available analysis kits (Sigma, USA) according to the manufacturerŌĆÖs direction. Glycolytic potential was calculated as glycolytic potential = 2├Ś([glycogen]+[glucose]+[glucose-6-phosphate])+[lactate] [18].
Measurements of enzyme activity
Hexokinase (HK) activity was determined using a commercial kit (Jiancheng Bioengineering Institute, Nanjing, China) based on the coupled reactions of hexokinase with glucose-6-phosphate dehydrogenase which resulted in the formation of reduced form of nicotinamide-adenine dinucleotide (NADH). One unit of HK was defined as the amount of enzyme to generate 1.0 mmole of NADH per minute at 37┬░C and pH 7.6. HK activity in muscle was expressed as HK units per gram of muscle protein (U/g protein).
Phosphofructokinase (PFK) activity was determined using an assay kit (Sigma, USA) by a coupled enzyme assay, in which fructose-6-phosphate and ATP were converted to fructose-1,6-diphosphate and ADP by PFK. The ADP was converted by the enzyme mix to AMP and NADH. One unit of PFK was defined as the amount of enzyme to generate 1.0 mmole of NADH per minute at 37┬░C and pH 7.6. PFK activity in muscle was expressed as PFK units per gram of wet muscle tissue (U/g muscle).
Glycogen phosphorylase a (GP) activity was determined using an assay kit (Baoman, Shanghai, China) based on the coupled reactions of GP with phosphoglucomutase and glucose-6-phosphate dehydrogenase which resulted in the formation of NADPH. One unit of GP was defined as the amount of enzyme to generate 1.0 ╬╝mole of G-1-P per minute at 30┬░C and pH 6.8. GP activity in muscle was expressed as GP units per gram of muscle protein (U/g protein).
Pyruvate kinase (PK) activity was measured using a commercial kit (Jiancheng Bioengineering Institute, Nanjing, China) based on the coupled reactions of PK with lactic dehydrogenase which resulted in the formation of NAD+. One unit of PK was defined as the amount of enzyme to transfer a phosphate group from PEP to ADP to generate 1.0 ╬╝mole of pyruvate per minute at 37┬░C and pH 7.6. PK activity in muscle was expressed as PK units per gram of muscle protein (U/g protein).
Western blotting
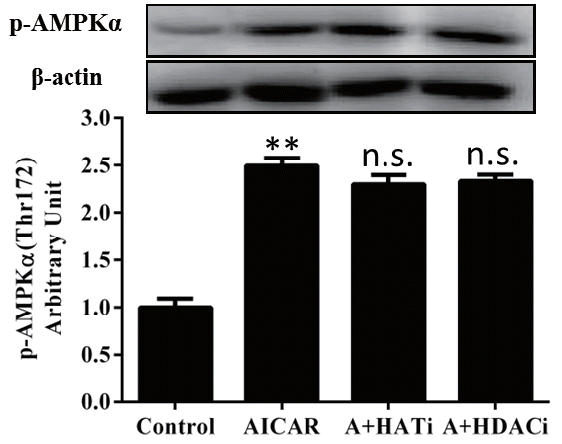
Frozen mouse LD muscle (0.05 g) was homogenized using a ULTRA TURRAX disperser (IKA, Staufen, Germany) in 300 ╬╝L of T-PER Tissue Protein Extraction Reagent (Thermo, Rockford, IL, USA ) containing 10 mM nicotinamide, 1 mM TSA and protease inhibitors (Roche, Basel, Switzerland). Each muscle homogenate was mixed with an equal amount of 2├Ś standard sodium dodecyl sulfate (SDS) sample loading buffer containing 0.5 M TrisŌĆōHCl (pH 6.8), 4.4% (w/v) SDS, 20% (v/v) glycerol, 2% (v/v) 2-mercaptoethanol, and 0.01% bromophenol blue, and boiled for 5 min before electrophoresis. A total of 40 ╬╝g proteins were loaded onto 12% gel and separated by SDSŌĆōpolyacrylamide gel electrophoresis (PAGE). The proteins on gels were then transferred onto PVDF membranes (Millipore, Billerica, MA, USA). The membranes were blocked at room temperature with 1% bovine serum albumin (BSA) in 1├Ś Tris-buffered saline-Tween 20 (TBST) for 1 h, incubated overnight at 4┬░C with rabbit acetylated-lysine antibody, anti-phospho-AMPK╬▒ (Thr 172) (Cell Signaling Technology, Beverly, MA, USA) or anti-╬▓-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted in 1% BSA/TBST. The membranes were washed 3├Ś in TBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma, USA) for 1 h at room temperature. Proteins were visualized by application of ECL chemiluminescent substrate (Bio-Rad, Hercules, CA, USA), imaged using the ChemiDoc XRS+ imaging system and quantified by using Quantity One software (Bio-Rad, USA) with ╬▓-actin as an internal reference. Band density among different blots was normalized according to the density of the reference band.
Statistical analysis
All data were subjected to analysis of variance as a completely randomized design using the general linear model (GLM) Procedure of Statistical Analysis System (SAS 9.1, 2002). Each animal was considered as an experimental unit for data analyses. Time course data were analyzed as a split-plot in time. Differences in the mean values among treatments at the same time point were compared by the DunnettŌĆÖs t test (p<0.05). Differences within the same treatments among different time points were compared by the FisherŌĆÖs Protected least significant difference (LSD) test (p<0.05). Mean values and standard errors of the means were reported.
RESULTS AND DISCUSSION
Inhibition of HAT and HDAC altered glycolysis in postmortem muscle
Glycolysis in postmortem muscle is supposed to follow the same reactions as in living animals. For this reason, the properties of the rate-limiting enzymes of GP, PFK, and PK have been comparatively studied in normal and PSE muscle in an effort to explain the aberrant glycolysis. Unexpectedly, many studies reported that the GP, PFK, and PK were not responsible for the altered metabolism in PSE muscle though increased activities were detected in some cases [19]. This indicates that the biochemistry of postmortem glycolysis is more complex than what has been expected and other regulatory mechanisms exist for fastened glycolysis in PSE muscle.
To test if reversible acetylation of ╔ø-amino groups on lysine residue plays a role in postmortem glycolysis, mice were intraperitoneally injected with AICAR, HAT, or HDAC inhibitors before slaughter and their effects on glycolysis in postmortem muscle were investigated. As expected, injection of AICAR significantly increased (p<0.01) the phosphorylation of AMPK╬▒ at Thr172 (Figure 1), indicating the increased AMPK activity. At the same time, activation of AMPK by AICAR decreased pH and increased lactate concentration (p<0.05) in LD muscle 45 min postmortem (Table 1). Although no difference was detected in muscle pH 24 h postmortem between AICAR and control mice, which could be contributed by proteolysis and nucleotide metabolism as the products of these processes, like ammonia and free amino acids, altered and could have eliminated the difference in muscle pH 24 h postmortem, higher lactate showed increased glycolysis in LD muscle from mice injected with AICAR. The altered glycolysis among treatments was not related to muscle glycogen storage since no difference in glycolytic potential was detected (Table 1). These data support previous studies that AMPK regulates glycolysis in postmortem muscle [7,8,20].
Antemortem injection of HAT inhibitor II diminished AICAR induced an increase in postmortem glycolysis. As shown in Table 1, injection of AICAR plus HAT inhibitor significantly increased (p<0.05) muscle pH postmortem when compared to that of mice injected with AICAR alone. At 24 h postmortem, the muscle pH of A+HATi mice became the highest, which was even higher than that of control mice. Consistent with muscle pH, the lactate content in postmortem muscle was decreased by HAT inhibitor II. Lactate in muscle from A+HATi mice was significantly lower (p<0.05) than that of AICAR mice both at 45 min and 24 h postmortem, which was statistically the same as in the muscle of control mice (Table 1). Opposite to HAT inhibitor, injection of HDAC inhibitors, TSA and NAM, in combination to AICAR further increased glycolysis in postmortem muscle of mice (Table 1). Among all the four treatments, the lowest pH and highest lactate concentration were both determined in the muscle from A+HDACi mice (Table 1). The influence of HAT and HDAC inhibitors on glycolysis was also evidenced by the altered glycogen, glucose and glucose-6-phosphate in postmortem muscle. Although no difference in glycogen content was determined early postmortem, higher glycogen (p<0.05) was determined 24 h postmortem in muscle from mice injected with HAT inhibitor among the three AICAR injected groups (Table 1), showing that the HAT inhibitor decreased the extent of glycolysis in muscle, which was consistent with the muscle pH values and lactate concentrations. Further, altered glucose (Table 1) and glucose-6-phosphate (Table 1) in postmortem muscles from mice injected with or without inhibitors illustrated that inhibitors regulated glycolytic enzyme activities and the glycolytic rate since glycolytic substrates were not different among mice at slaughter (Table 1). The altered metabolism in postmortem muscle induced by HAT and HDAC inhibitors suggested that, first, HAT and HDAC, or reversible protein acetylation was involved in postmortem glycolysis; second, protein acetylatoin mediated AMPK regulation of postmortem glycolysis though further proof is needed.
Protein acetylation was involved in AMPK regulation of postmortem glycolysis
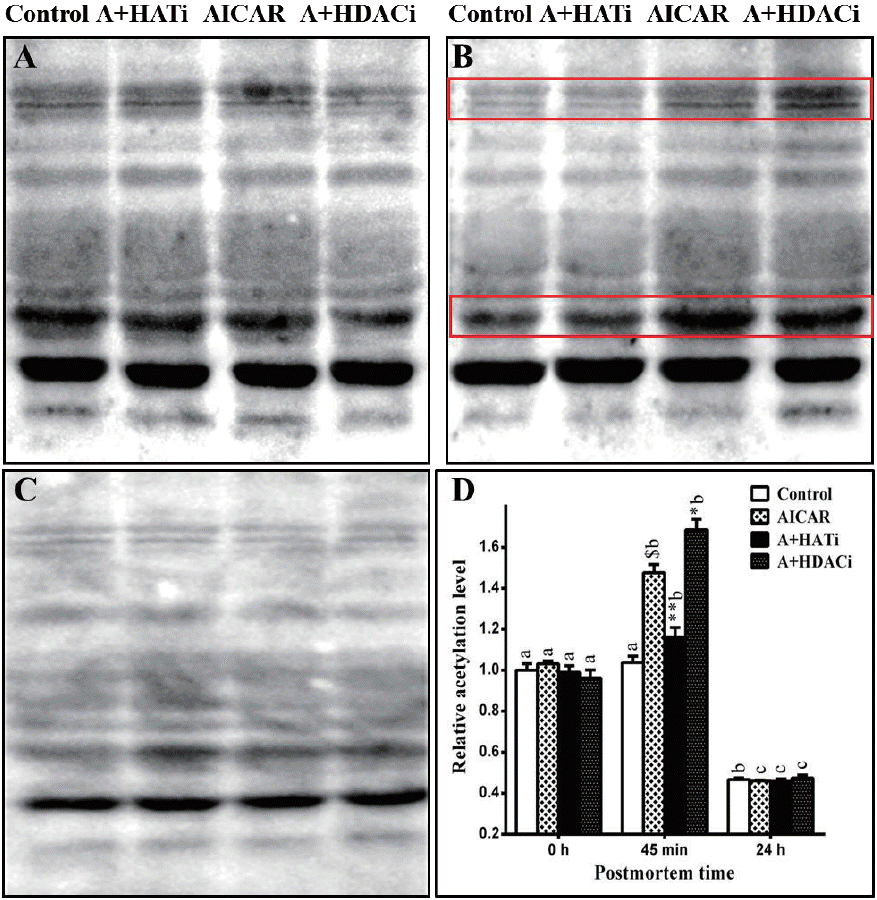
Protein acetylation in muscle was evaluated by western blotting. As shown in Figure 2, total acetylated proteins did not vary early postmortem in the control muscle, but decreased significantly 24 h later (p<0.05), indicating increased protein deacetylation late postmortem. Injection of AICAR increased (p<0.05) the total amount of acetylated proteins in muscle 45 min postmortem when compared to that at slaughter, showing that AMPK activation up-regulated protein acetylation in muscle early postmortem. A day later, the total acetylated proteins also decreased in the AICAR muscles, which became lower than those at 0 h postmortem (p<0.05).
No difference in total acetylated proteins among the four treatments was determined at 0 h (Figure 2). Injection of AICAR significantly increased (p<0.05) the total acetylated proteins in muscle 45 min postmortem when compared to that of the control. As expected, injection of HAT inhibitor diminished the up-regulation of protein acetylation by AMPK activation and HDAC inhibitors further increased (p<0.05) the total acetylated proteins in muscle 45 min postmortem when compared to that of the AICAR treatment. For all four treatments, the total acetylated proteins decreased and reached the lowest values 24 h postmortem. Immunoblotting of acetylated proteins in muscle 45 min postmortem showed that at least three protein bands were obviously influenced by AICAR, HAT, and HDAC inhibitors (as shown within red boxes). Further studies are needed to identify these proteins.
Although it is well established by many studies that AMPK regulates glycolysis in postmortem muscle, the underlying mechanisms are not well defined. In ischemic heart and hypoxic skeletal muscle, activated AMPK increases glycolysis by two signaling pathways. First, AMPK phosphorylates and activates phosphorylase kinase, which then phosphorylates and activates glycogen phosphorylase, an enzyme controlling glycogenolysis and catalyzing the production of substrate for glycolysis [21]. Second, AMPK regulates glycolysis by phosphorylating and activating phosphofructokinase-2 (PFK-2) [22]. In mice, activation of AMPK by AICAR did not increase glycogen phosphorylase activity in postmortem muscle [7]. In pigs, higher AMPK activity was detected 30 min postmortem in the muscle that became PSE meat, the muscle of halothane carriers, and the muscle of antemortem stressed pigs, but the glycogen phosphorylase activity was not altered in these muscles early postmortem [17,23,24]. In addition, the structural and kinetic characteristics of glycogen phosphorylase from normal versus PSE muscles, normal versus halothane sensitive pigs were not different [25]. All these studies suggest that glycogen phosphorylase does not mediate AMPK induced increase of glycolysis in postmortem muscle and is not responsible for the development of PSE meat. Indeed, higher fructose-2,6-biphosphate has been detected in the muscle that became PSE meat [17], but no direct evidence has proved that AMPK regulates PFK in postmortem muscle.
Here the influence of AMPK activation on protein acetylation was investigated in a relationship to glycolysis in postmortem muscle. Activation of AMPK by AICAR increased postmortem glycolysis as proved by lower pH and higher lactate concentration in muscle (Table 1). At the same time, it significantly increased the total acetylated proteins in muscle 45 min postmortem (Figure 2). Antemortem injection of HAT inhibitor eliminated the effect of AMPK activation on protein acetylation (Figure 2), which simultaneously diminished the effect of AICAR on glycolysis in postmortem muscle (Table 1). Inhibition of HDAC by TSA and NAM further increased protein acetylation from 0 to 45 min postmortem in a combination to AMPK activation (Figure 2), which also led to the lowest pH and highest lactate concentration in postmortem muscle (Table 1). The inverse relationship between muscle pH and total acetylated proteins, but a positive correlation between lactate accumulation and total acetylated proteins in muscle 45 min postmortem supported that protein acetylation was involved in AMPK regulation of postmortem glycolysis.
The total acetylated proteins in muscle from control mice did not increase but tended to decrease postmortem. Activation of AMPK increased the total acetylated proteins in muscle 45 min postmortem, which then decreased and reached the lowest level 24 h postmortem (Figure 2). As AMPK mediates the effect of pre-slaughter stress on postmortem glycolysis [23], it is logical to deduce that protein acetylation is a mechanism by which antemortem stress influences postmortem metabolism and meat quality. This is supported by the observation that forced swimming antemortem increased AMPK activity, the total acetylated proteins and lactate content, and decreased pH in muscle 45 min postmortem [26]. Identification and characterization of the differentially acetylated proteins (Figure 2) is necessary for further insight into this rationale.
Glycolytic enzymes in postmortem muscle
Due to the technical challenge to identify acetylation sites, most studies on protein acetylation have been focused on histones and nuclear proteins [27,28]. Recently, the development of acetylated lysine antibody along with high throughput proteomics has greatly facilitated the research in protein acetylation. More and more non-histone proteins have been identified. Totally, more than 2000 proteins in mammalian cells have been reported to be acetylated, which include all the enzymes involved in glycogenolysis and glycolysis except glucophosphomutase and phosphoglucose isomerase [29]. GP, HK, PFK, and PK are rate limiting enzymes in glycolysis that are modified by acetylation, but the impact of acetylation on their function is unclear [13,29].
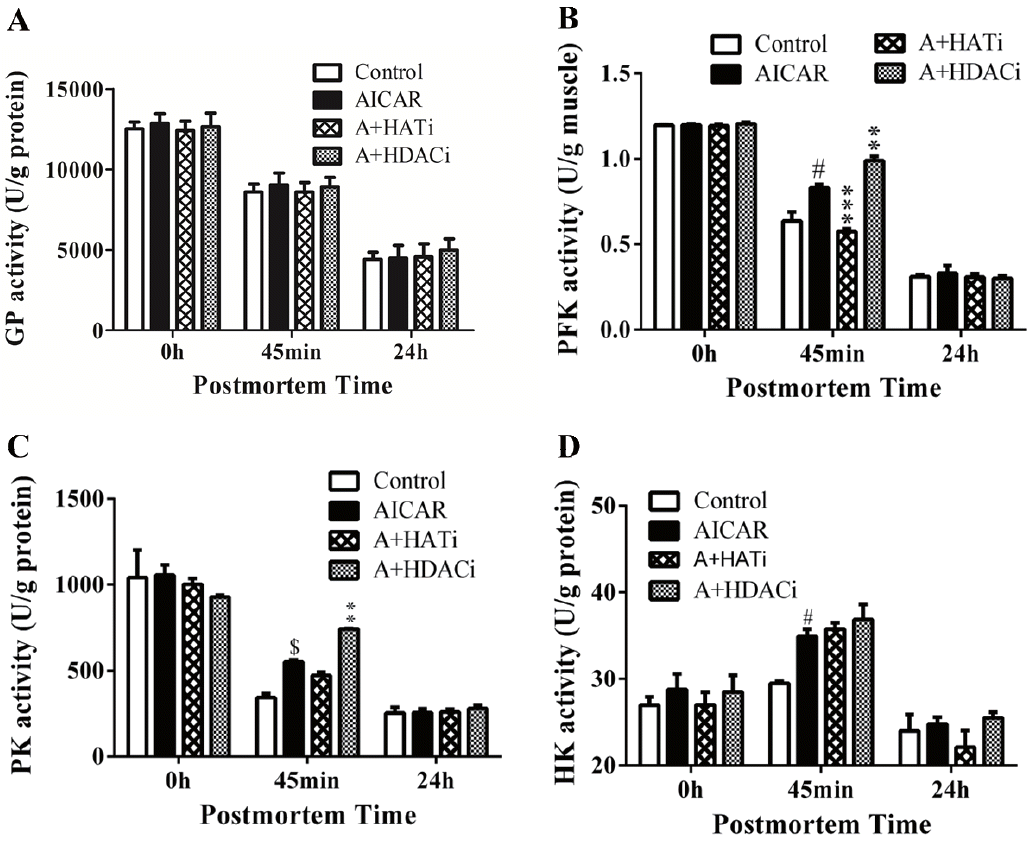
In consistence with literature [7], antemortem injection of AICAR did not change GP activity in mouse postmortem muscle (Figure 3A). However, similar to muscle pH and lactate concentration, the PFK activity was altered by the injection of AICAR, HAT, and HDAC inhibitors (Figure 3B). When compared to control, activation of AMPK by AICAR significantly increased PFK activity in muscle 45 min postmortem (p<0.05). Injection of HAT inhibitor II inhibited AICAR induced protein acetylation in muscle 45 min postmortem (Figure 2), which decreased PFK activity as well (p<0.001). On the contrary, HDAC inhibitors, TSA and NAM, further increased the amount of total acetylated proteins in combination to AICAR in muscle early postmortem (Figure 2), which also led to the highest PFK activity in A+HDACi group. As PFK can be modified by acetylation, the altered PFK activity by HAT and HDAC inhibitors suggests that protein acetylation may regulate PFK activity.
As previously reported [7], PK activity was increased by AICAR in mouse muscle early postmortem, which was further increased by HDAC inhibitor when injected with AICAR (Figure 3C), suggesting that protein acetylation/deacetylation might be involved in regulating PK activity. Different to PFK, injection of HAT inhibitor did not decrease PK activity when compared to the AICAR muscle. The different response of PFK and PK to HAT inhibitor II may reflect their distinct regulation mechanisms. In addition, higher HK activity was detected early postmortem in muscle from mice injected with AICAR (p<0.05), but HAT and HDAC inhibitors showed no effect on its activity (Figure 3D).
In summary, protein acetylation may play an important role in the regulation of glycolysis in postmortem muscle. AMPK activation increased the total acetylated proteins in muscle early postmortem. Several differentially acetylated protein bands have been detected in muscles with different glycolytic rates. Identification and characterization of these differentially acetylated proteins will provide new insight into the biochemistry of postmortem glycolysis.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print







