Metagenomic investigation of gastrointestinal microbiome in cattle
Article information
Abstract
The gastrointestinal (GI) tract, including the rumen and the other intestinal segments of cattle, harbors a diverse, complex, and dynamic microbiome that drives feed digestion and fermentation in cattle, determining feed efficiency and output of pollutants. This microbiome also plays an important role in affecting host health. Research has been conducted for more than a century to understand the microbiome and its relationship to feed efficiency and host health. The traditional cultivation-based research elucidated some of the major metabolism, but studies using molecular biology techniques conducted from late 1980’s to the late early 2000’s greatly expanded our view of the diversity of the rumen and intestinal microbiome of cattle. Recently, metagenomics has been the primary technology to characterize the GI microbiome and its relationship with host nutrition and health. This review addresses the main methods/techniques in current use, the knowledge gained, and some of the challenges that remain. Most of the primers used in quantitative real-time polymerase chain reaction quantification and diversity analysis using metagenomics of ruminal bacteria, archaea, fungi, and protozoa were also compiled.
INTRODUCTION
The rumen microbiome is a complex community of prokaryotes, eukaryotes, and viruses. The prokaryotes include bacteria, primarily anaerobic bacteria, and archaea, primarily methanogens. Anaerobic fungi and protozoa, exclusively ciliates, constitute the eukaryotic community. The large number of species present and their uneven distributions among different species create several challenges to analyze or characterize any rumen microbiome comprehensively. The inability to culture most microbes in the rumen prompted development and application of cultivation-independent methods and technologies starting in the 1990’s (reviewed by [1–3]). Those approaches and technologies are primarily based on the 16S or 18S rRNA genes as the phylogenetic markers. Hybridization using specific oligonucleotide probes (including fluorescent in situ hybridization) (e.g., [4,5]), differential migration of DNA fragments in denatured gel matrices (including denaturing or temperature gradient gel electrophoresis and ribosomal intergenic spacer analysis) (e.g., [6–9]), and DNA sequencing based on the Sanger sequencing technology (e.g., [10,11]) were the primary methods. Studies using one or more of these methods allowed detection or identification of many microbes that had not been known to be present in the rumen and improved our understanding and appreciation of the diversity and complexity of the rumen microbiome. The global diversity framework of the rumen bacteria and archaea was established based on the sequence data produced in a large number of studies that use the Sanger sequencing technology [12]. These methods and technologies were replaced about 10 years ago by the next-generation DNA sequencing technologies.
Metagenomics empowered by the so-called next-generation sequencing (NGS) was first used to reveal the glycoside hydrolases in the rumen of dairy cows [13] and to evaluate the diversity of bacteria in the rumen and feces of beef cattle [14]. By now, hundreds of metagenomic studies have been reported to address different aspects of the rumen microbiome, such as the effect of feed additive or diets on the rumen microbiome, early patterns of colonization of young rumen, and the diversity of the enzymes, especially glycoside hydrolases. In this review, we discussed metagenomic investigation of gastrointestinal (GI) microbiome in cattle using quantitative real-time polymerase chain reaction (qPCR), phylogenetic microarrays, and NGS. The limitations and future perspectives of metagenomic studies of GI microbiome are also discussed.
SAMPLING AND DNA EXTRACTION
Rumen cannulation has been used as a standard method to facilitate sample collection from the rumen of cattle [15]. However, only a small number of ruminally cannulated cattle are available to any researcher in a single study, thus limiting the number of livestock that can be sampled to evaluate the ruminal microbiome if only rumen cannulated cows are used. Rumen contents can be collected from non-cannulated cattle using stomach tubing, and any number of cattle can be sampled. A previous study showed that these two different methods did not result in significant difference in the composition of ruminal microbiome [15], indicating that stomach tubing can be used as an alternative method to collect ruminal samples from a large number of cows that are required by the investigators. However, this approach cannot be used to repeatedly collect rumen samples from the same cattle within a short period (e.g., a day) because the cattle are stressed during the sampling. Contamination by saliva may not affect the results of microbiome analysis, but it can alter the fermentation characteristics. Thus, precaution needs to be taken to minimize saliva contamination.
Metagenomic DNA can be extracted from the collected rumen contents. The rumen contents can also be separated into a liquid fraction and a solid fraction if desired and then subjected to separate metagenomic DNA extraction as described previously [6]. The separation of the two fractions allows analysis and identification of the microbes present in each of the fractions. Fresh fecal samples can be collected from cattle by rectal grab with a glove [16], while samples from other GI segments, such as cecum and jejunum, can only be collected after cattle are harvested [17,18].
Because different microbes are present in GI samples, metagenomic DNA representing the entire microbiome needs to be extracted. It has been reported that bead-beating based methods are more efficient than other methods to extract metagenomic DNA from GI samples [19]. Yu and Morrison [19] developed the repeated bead beating plus column (RBB+C) purification method to efficiently extract metagenomic DNA from ruminal and other GI samples, and this method increased DNA yield by up to 6-fold compared to three other common methods. The RBB+C method has been widely used to extract metagenomic DNA from ruminal and lower GI samples in cattle and has been cited for more than 660 times (e.g. [17,18,20–24]). It should be noted that many studies used commercial general purposed DNA extraction kits to extract metagenomic DNA from GI samples. Because most of those commercial kits were developed for general purposes, they may not yield representative DNA, thus likely skewing the actual composition and structure of GI microbiomes.
PHYLOGENETIC MARKER GENES
The 16S rRNA genes have been used as the marker gene to determine the composition of both bacterial and archaeal communities because it is phylogenetically conserved and it does not laterally transfer [25]. The 16S rRNA gene sequence has a characteristic organization, with 9 hypervariable regions separating 10 conserved regions (Figure 1) [9,26]. The sequences of the conserved regions are similar (the degree of similarity depends on the phylogenetic relatedness) among bacterial or archaeal species, and they can be used to identify most bacteria or archaea. The conserved regions (C1–C9) are used to design PCR primers to amplify a defined portion of the 16S rRNA gene. The 16S rRNA gene sequences of the hypervariable regions (V1–V9) are different among bacterial or archaeal species and can be targeted to identify individual bacterial or archaeal species using PCR with species-specific primers for the 16S rRNA gene. Universal and species-specific PCR primer sets that are commonly used in examining microbial populations are listed in Table 1 and 2. The resultant 16S rRNA gene amplicons can be subjected to analyses using clone library, denaturing gradient gel electrophoresis (DGGE), qPCR, phylogenetic microarray, and NGS. In addition to 16S rRNA genes, methyl coenzyme-M reductase subunit A, that is unique of methanogens, has been used as a phylogenetic marker gene to examine methanogen populations [27,28] including the ruminal methanogen populations (e.g. [29–32]).

A diagram showing the alternate conserved regions and hypervariable regions of the 16S rRNA genes. C1–C10, the 10 conserved regions; V1–V9, the 9 hypervariable regions. Alignments with high sequence similarity are highlighted in black, whereas alignments with low sequence similarity are highlighted in gray.

Primers used for quantitative real-time polymerase chain reaction analysis of ruminal bacteria

Primers used for quantitative real-time polymerase chain reaction of ruminal archaea, protozoa, and fungi
META-ANALYSIS OF MICROBIAL DIVERSITY
Cloning of 16S rRNA gene amplicons and then Sanger sequencing were the primary method to reveal the composition of GI microbiome in cattle before NGS became affordable about 7 years ago. Some studies also sequenced excised DGGE band using Sanger sequencing to identify the bacteria of interest. Most studies using these methods focused on the analysis of bacterial communities in the liquid fraction of the rumen (e.g., [33–36]), and only a small number of studies analyzed bacterial communities in the solid fraction of the rumen (e.g., [6,13]). Some studies examined bacterial communities present on the rumen wall [37,38]. Although these methods are no longer commonly used these days, they can still help identify novel bacteria [39]. Additionally, when genus-specific primers are used, the diversity of a specific genus such as Prevotella and Treponema in the rumen can be examined [40,41].
All the 16S rRNA gene sequences obtained using cloning and Sanger sequencing have been archived in GenBank, which stores all the sequences including sequences of poor quality and chimeric sequences from any sample. The ribosomal database project (RDP, http://rdp.cme.msu.edu/) archives all 16S rRNA gene sequences that passed quality controls. A meta-analysis of 16S rRNA gene sequences that are publicly available in the RDP database can be used to investigate GI microbiome in cattle (Figure 2). Kim et al [12] first retrieved all the 16S rRNA gene sequences of both ruminal bacteria and archaea deposited in the RDP database and then conducted a meta-analysis of the retrieved sequences to examine the collective bacterial and archaeal diversity in the rumen. In that study, a total of 13,478 bacterial and 3,516 archaeal 16S rRNA gene sequences were obtained from the RDP database (Release 10, Update 22). Those sequences were used to establish a framework of bacterial and archaeal diversity in the rumen [12]. Firmicutes and Bacteroidetes were predominant bacterial phyla, while Euryarchaeota was the most predominant archaeal phylum. A recent study [42] updated the appraisal of the ruminal archaeal diversity using a meta-analysis of 8,623 archaeal sequences retrieved from the RDP database (Release 11, Update 3).

A flowchart for meta-analysis of gastrointestinal (GI) microbial diversity in cattle.
A similar meta-analysis was also performed to assess the composition of bacterial communities in the feces of cattle [43]. In this study, a total of 13,663 bacterial sequences recovered from the feces of cattle were retrieved from the RDP database (Release 11, Update 1). Firmicutes and Bacteroidetes were the predominant phyla in the feces of cattle, but the fecal and ruminal microbiome differed in the membership of these two major phyla.
QUANTITATIVE REAL-TIME POLYMERASE CHAIN REACTION
The qPCR has been used to accurately quantify GI microbial populations in cattle [44]. By continued efforts of rumen microbiologists in the past two decades, various pairs of universal and specific primers have been developed and validated using rumen samples. These primers support not only detection but also quantification of various groups of rumen microbes. In this review we compiled most of the primers used in the qPCR analysis of bacteria (Table 1) and archaea, protozoa, and fungi (Table 2). Compared to the other methods, qPCR is sensitive and quantitative, allowing reliable analysis of individual microbial populations, with respect to population dynamics and responses to dietary interventions. It should be noted that although qPCR does not allow quantification of numbers of microbial cells per unit of the sample, the gene copy numbers determined by qPCR is a reliable measure of the abundance of a microbial population.
Domain-specific primer sets for the 16S rRNA gene have been designed to detect and quantify all bacteria or archaea, while phylum-specific primer sets were designed to target members of a phylum (Table 1). Genus-specific primers are frequently used to determine the abundance of a genus such as Prevotella, Bacteroides, Butyrivibrio, or Ruminococcus (Table 1). Studies using qPCR have provided interesting insight into abundance and thus the importance of some ruminal bacteria. For instance, among the known bacterial genera, Prevotella was the most predominant regardless of diet or location in the rumen [45,46]. However, the three cultured species of this genus, P. ruminicola, P. bryantii, and P. brevis, each accounts for only less than 3.8% of Prevotella, suggesting that new species remain to be identified and characterized within the genus Prevotella [45–47].
Tajima et al [48] designed 12 primer sets specific for culturable ruminal bacterial species that include Fibrobacter succinogenes, Ruminococcus flavefaciens, Ruminococcus albus, Prevotella ruminicola, Prevotella albensis, Prevotella bryantii, Selenomonas ruminantium-Mitsuokella multiacida, Streptococcus bovis, Eubacterium ruminantium, Treponema bryantii, Succinivibrio dextrinosolvens, and Anaerovibrio lipolytica. These ruminal bacterial species as affected by diet shifts were monitored using the specific primers. Populations of rumen Megasphaera elsdenii, Butyrivibrio fibrisolvens, and Streptococcus bovis also were monitored in cattle fed different diets using qPCR [49,50]. Additionally, qPCR was also used to quantify uncultured ruminal bacteria represented by new 16S rRNA gene sequences [45,51], providing opportunities to help understand the importance and functions of uncultured microbes in GI environment.
The primer sets specific for total archaea and their lower taxa (order, family, and genus) were listed in Table 2. Among ruminal methanogens, Methanobrevibacter has been identified as the most dominant genus [12]. Recently, the shift of metabolically active methane producers by many of the dietary interventions has been confirmed by qPCR. The “rumen cluster C” (RCC), now placed in the new order Methanoplasmatales, was suggested as a potential methane mitigating target due to its significant positive correlation with reduced methane upon oil supplementation [52].
The procedure developed by Sylvester et al [53] has been widely used to quantify the abundance of protozoa in the rumen (Table 2), and the qPCR result was comparable to that of microscopic counting, which needs expertise and experience to identify genus or species of ruminal protozoa. This primer pair had been used in quantifying the abundance of total ruminal protozoa in various studies [54–56]. Primers targeting the 18S rRNA genes of specific taxa of ruminal protozoa, including Entodinium spp. and Dasytricha ruminantium, have also been developed and used in quantifying individual populations of ruminal protozoa [57].
Anaerobic fungi are not a large group of microbes in the rumen, but their hyphae can penetrate into the fiber helping colonization by other cellulolytic bacteria [58]. Specific primers for ruminal anaerobic fungi were first reported by Denman et al [59] (Table 2). Genera Neocallimastix, Orpinomyces, and Piromyces were successfully amplified by the primer set without amplification of other aerobic fungal genera. Most fungal primers were designed to amplify the internal transcribed spacer 1, rather than the fungal 18S rRNA gene because the latter is too conserved to provide an adequate phylogenetic resolution [60]. Another Neocallimastigales-specific qPCR primer set targeting the 5.8S rRNA gene was first validated by Edwards et al [61], but few studies have ever used it.
Several recent papers investigated the bacterial communities in the lower GI tract of pre-weaned calves and steers using qPCR [62,63], and these studies advanced our limited knowledge about the microbiome residing in the small and large intestines and its roles in post-ruminal feed digestion. The composition of methanogen communities appeared to differ not only among different segments of the intestines (jejunum, ileum, cecum, colon, and rectum) but also among individual animals [64]. Frey et al [65] detected methanogens in the duodenum of lactating dairy cows, but it remains to be determined if they are live methanogens.
PHYLOGENETIC MICROARRAY
A phylogenetic microarray is a 16S rRNA gene chip composed of a large number of oligonucleotide probes. It can simultaneously detect predominant microbes in a complex ecosystem [44]. The phylogenetic microarray technique has been widely used to investigate the human GI microbiome [66,67]. The first microarray dedicated to the analysis of ruminal microbiome, referred to as RumenBactArray, was developed by Kim et al [68]. The RumenBactArray was developed using the 16S rRNA gene sequences that were obtained for a meta-analysis of rumen microbial diversity [12]. In total, 1,666 specific microarray probes were included in the RumenBactArray, and six of these arrays can be synthesized on each chip, allowing simultaneous analysis of six samples, with each probe being in three replicates per array. Specificity of the microarray probes was evaluated using known 16S rRNA gene clones obtained in a previous study [39]. The utility of the RumenBactArray was tested using liquid and solid fractions of rumen samples from sheep fed two different diets [51]. The structure of ruminal bacterial communities was greatly different between the two diet groups and between the two fractions in the same diet group. This RumenBactArray was recently used to examine ruminal bacterial communities as affected by essential oils intended to decrease methane production [69]. The RumenBactArray analysis revealed bacterial population shifts upon exposure to the essential oils.
Although phylogenetic microarrays can rapidly assess changes in microbial populations in a microbiome, they have some limitations. The microarray method is not as quantitative, sensitive, or specific as qPCR, and it can only detect dominant microbial populations [44]. Additionally, the microarray method cannot detect novel microbial populations that are not represented by the probes because only known sequences are used for probe design. New probes need to be added to overcome this limitation. Another technical limitation is that poor probe design and hybridization can result in inaccurate profiles of microbial populations. Sensitivity involved in probe hybridization is positively associated with probe length, whereas the opposite holds true for specificity [70]. To improve both sensitivity and specificity, Kim et al [68] used the GoArray program [71] to design the RumenBactArray probes, each of which is composed of 2 sub-probes, and a linker between the 2 sub-probes. Additionally, the specificity of microarray probes can be affected by the number of reference sequences used in the design of probes. Therefore, the specificity of microarray probes will need to be regularly checked based on updated reference sequences.
The RumenBactArray was developed when 454 pyrosequencing was the only NGS available for metagenomic analysis. Now that Illumina systems, especially the MiSeq platform, allow cost-effective sequencing-based metagenomic analysis, most people use NGS. However, because microarray can provide a uniformed analysis and microarray data are easier to analyze, microarrays may become a useful tool once the composition of rumen and GI microbiome of cattle has been determined.
NEXT-GENERATION SEQUENCING
Since the 454 Genome Sequencer FLX system developed by 454 Life Sciences (Roche, Branford, CT, USA) was used for 16S rRNA gene amplicon sequencing, microbiomes in various environments have been investigated. Brulc et al [13] first investigated the ruminal bacterial communities of cattle using 454 pyrosequencing and revealed the microbial difference between liquid and adherent fractions. Since then, many studies have examined the ruminal bacterial communities of cattle using 454 pyrosequencing by analyzing the V1–V3 region of the 16S rRNA genes (Table 3). These studies showed that ruminal microbiome is greatly affected by diets and feed additives (e.g., [14,20,21,72]). In addition, Cersosimo et al [73] analyzed the composition of ruminal methanogens using 454 pyrosequencing of archaeal 16S rRNA gene amplicons generated from a pair of primers specific for methanogens, while Fouts et al [74] investigated the ruminal fungal diversity using a fungal-specific 18S rRNA primer pair (Table 3). Ruminal protozoal communities also were examined using a protozoal-specific 18S rRNA primer pair [75]. Due to the high cost and low throughput, 454 pyrosequencing is no longer used in analyzing any microbiome.

Ribosomal RNA gene primer pairs used for the next-generation sequencing method
Compared to the 454 pyrosequencing, the Illumina sequencing technique is more cost-efficient and has much greater throughput. Caporaso et al [76] developed a workflow to examine bacterial diversity using the Illumina GAIIx platform by analyzing the V4 region of the 16S rRNA genes (Table 3). However, in the past few years, the Illumina MiSeq system has been the primary technology for microbiome analysis (Illumina, San Diego, CA, USA). The MiSeq system can produce 500 bp sequence reads by using the 2×300 bp paired-end sequencing that is comparable to those generated by 454 pyrosequencing. The V1–V3 region [23] and the V4–V5 regions [77] have been the primary regions sequenced using the Illumina MiSeq platform when ruminal bacterial communities are analyzed. Table 3 lists most of the primers that have been used to generate PCR amplicons for NGS sequencing. The Illumina MiSeq platform has also been used to analyze microbiome in the lower GI tract (cecum, jejunum, and colon) of cattle [17,18,22,24], and Firmicutes was found to be the largest phylum in these intestine segments [17,18,22,24]. It is also shown that the rumen and the lower GI tract harbor phylogenetically distinct microbiome [43].
The Pacific Biosciences (PacBio) sequencing is considered one of the third generation of DNA sequencing technologies (Pacific Biosciences, Menlo Park, CA, USA). Only one study has been reported that used this technology to analyze the ruminal microbiome [78]. Using the PacBio RSII system, nearly full-length (V1–V8) 16S rRNA genes can be sequenced (Table 3), providing finer phylogenetic resolution. The PacBio RSII system and the MiSeq system resulted in different microbiome composition and structure. Future studies are needed to determine which technology can support more accurate characterization of rumen and GI microbiome. A model or mock microbiome with known composition and structure will be required for such a comparison.
Specialty bioinformatics programs are required to analyze the large volumes of NGS data. QIIME [79] and Mothur [80] are the commonly used bioinformatics software packages to analyze 16S rRNA gene sequences generated from NGS technologies. A flowchart summarizing the bioinformatics process is shown in Figure 3. First, the NGS datasets are preprocessed to check sequence quality and to remove potential chimeric sequences and low-quality (often Q<20) sequences. ChimeraSlayer [81] and UCHIME [82] are widely used to detect and remove possible chimeric sequences. The cleaned sequences are clustered into operational taxonomic units (OTUs) using one of three OTU picking methods that are de novo, closed-reference, and open-reference methods based on a sequence similarity value (often 97%). The de novo method does not use a reference sequence set but clusters sequences into OTUs based the chosen sequence similarity value. In de novo OTU picking, all sequences are clustered into OTUs. Both the closed-reference and the open-reference OTU picking methods use a reference sequence core set. When the closed-reference OTU picking method is used, sequences are clustered against a reference sequence set based on the chosen sequence similarity value. Sequences that do not match any reference sequences are excluded. The open-reference OTU picking process does closed-reference OTU picking first, and the sequences that do not match a reference of the reference sequence set are clustered into OTUs de novo. Uclust is the default OTU clustering tool to pick OTUs from unaligned sequences in QIIME [79,83], while furthest neighbor, average neighbor, or nearest neighbor algorithm is used to generate OTUs from aligned sequences in Mothur [80]. Each OTU can be assigned to a taxon by comparing its representative sequence to the reference sequences in specialty databases such as Greengenes [84,85] and Silva [86–88]. A commonly used taxonomic assignment program is the Naïve Bayesian Classifier [89], which is the default method in both QIIME [79] and Mothur [80].
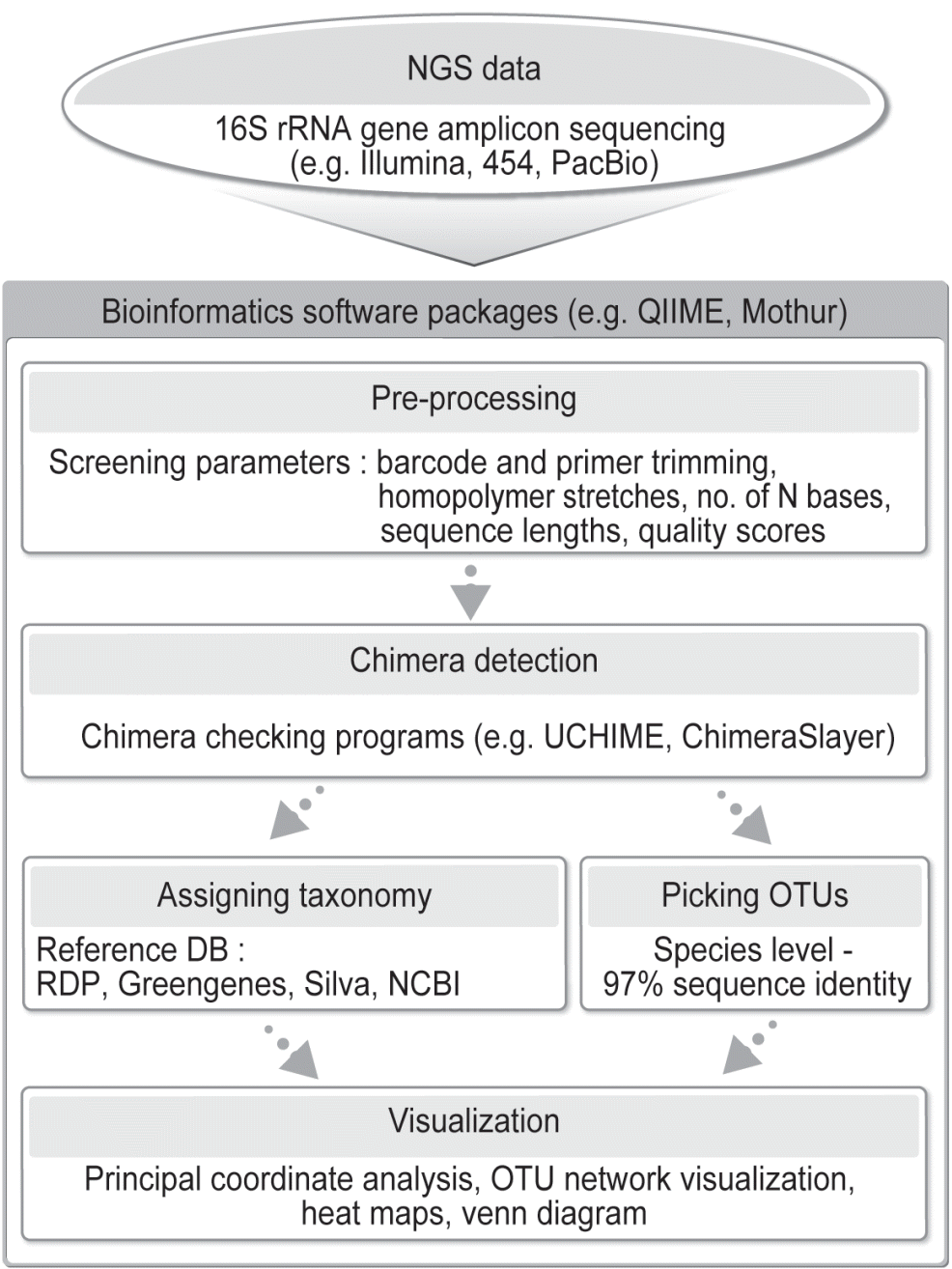
A flowchart outlining the process of bioinformatic analysis of 16S rRNA gene amplicons sequenced using next-generation sequencing (NGS). Either QIIME or Mothur can be used.
Alpha diversity measures the diversity in a given sample, while beta diversity measurements allow comparison of diversity among samples. For analysis of microbiome, OTUs at 97% sequence similarity are regarded as species equivalent and are used for both alpha and beta diversity statistics. Examples of alpha diversity analysis are observed richness (number of detected OTUs) and predicted richness (rarefied, Chao1, and ACE estimates of maximum species richness). On the other hand, beta diversity comparison is typically performed using multivariate analyses such as principal coordinates analysis [90], partial least square discriminant analysis, canonical correspondence analysis (CCA) [91], analysis of similarities [92], and permutational analysis of variance [93]. All these analyses allow visualization of similarity or dissimilarity among samples, while CCA also visualizes correlations between samples and environmental parameters.
Determination of the composition and structure of the rumen and the GI microbiome is important, but this type of microbiome has high functional redundancy [94], and it has been shown that phylogenetic variations in the human gut microbiome are not necessarily accompanied by functional changes [95]. Therefore, it is important to characterize rumen and GI microbiome with respect to its functional diversity and features. Phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) is a bioinformatics tool that has been used to predict metagenomic functions using 16S rRNA marker gene sequences [96]. Liu et al [97] used PICRUSt to examine the potential functionality of ruminal bacterial communities. However, members of the same species may have very different functions. This is exemplified by some strains of Escherichia coli and Ruminococcus albus. Therefore, it should be cautious to interpret the functional profiles predicted from 16S rRNA gene sequence using PICRUSt.
Although the 16S rRNA gene-based NGS methods have greatly contributed to the appraisal and understanding the diversity of rumen and GI microbiome, the resultant microbiome diversity may not be accurate due to limitations of PCR, such as differences in amplification efficiency, nonspecific annealing, and PCR artifact formation [98–100]. Thus, caution should be exercised when NGS data are interpreted quantitatively.
Shotgun metagenomic sequencing is one of the primary technologies to investigate the functional profiles of microbiomes [101]. Shotgun metagenomic sequencing empowered by NGS can potentially identify all the genes in GI microbiome in cattle. Several studies have used shotgun sequencing to investigate the functional diversity in the ruminal microbiome of cattle, including cattle fed methane-mitigating diets [102], cattle fed different forages [103], and cattle with wheat-induced frothy bloat [104]. This approach has also been used in comparatively characterizing the functional profiles in the fecal microbiome of buffaloes and cattle [105]. The advantage of shotgun metagenomic sequencing is that it does not have potential PCR biases of the 16S rRNA gene-based NGS method [99]. One limitation of the shotgun sequencing approach is that it can only reveal the functional potential, not the actual functions expressed in any microbiome.
Metatranscriptomics allows direct sequencing of the RNA transcripts expressed in a microbiome. Therefore, metatranscriptomics is better suited than shotgun metagenomic sequencing in functional analysis of rumen microbiome. RNA-Seq is the preferred technology to perform metatranscriptomic analysis of rumen microbiome. However, because more than 80% of the total RNA is rRNA, efficient functional profiling of rumen microbiome requires removal of the rRNA from the total RNA preparation. Several commercial kits, such as the Ribo-Zero rRNA Removal Kit (Bacteria) (Illumina, USA), MICROBExpress Bacterial mRNA Enrichment Kit (Ambion, Austin, TX, USA), RiboMinus Transcriptome Isolation Kit (Life Technologies, Gaithersburg, MD, USA), and NEBNext rRNA Depletion Kit (New England Biolabs, Beverly, MA, USA), are available to achieve a reasonably adequate depletion of rRNA. Because of the difficulty in isolating high-quality RNA and depletion of rRNA, metatranscriptomics has just been used in a few studies, including the study that characterized the functional profiles of the consortia adherent to the bovine rumen [106,107]. As the sequencing cost continues to fall and the output of sequencing technologies continues to increase, total RNA may be sequenced directly without depleting the rRNA. Total RNA sequencing will provide the opportunity to characterize both the composition of the functions of a microbiome. If the 16S rRNA genes are also sequenced, the ratio of rRNA over 16S rRNA gene can be used to assess the metabolic activity status of a bacterial taxon [108].
From a nutritional perspective, it is useful to identify the species or taxa of microbes that contribute to important aspects of rumen functions, such as fiber digestion, proteolysis, and rumen acidosis. Even though we know the metabolism and the role of some rumen microbes, it remains to be difficult, if not impossible, to ascertain the causality relationship between a group of microbes and a rumen function. In recent studies, correlation analysis has been used to identify the microbes that are correlated, either positively or negatively, to given fermentation characteristics or animal performance (e.g. [23,109]). Although causality relationship cannot be identified from such correlations, the correlated microbes can serve as indicators of fermentation or animal performance. New hypotheses can be formulated to further investigate if the correlated microbes are the cause of phenotypes of interest.
CONCLUDING REMARKS
Globally, the demands for beef and dairy products continue to grow as the population grows and the living standards improve in developing countries. Numerous nutritional studies have been conducted to improve feed efficiency and host health, and investigation and analysis of rumen microbiome, mostly using metagenomics, are increasingly being included in nutritional studies. Metagenomic studies of rumen microbiome may not directly enhance nutrition or host health, but they will provide the knowledge to help develop new rational dietary interventions to improve feed efficiency and host health. Furthermore, the density of microbes in the rumen is among the highest, creating intense interactions among microbes, both among the members of the same species or different species, including interactions between bacteria and phages. Metagenomic studies of the rumen microbiome will advance understanding of microbiology, especially microbial interactions, fermentative metabolism, and interspecies hydrogen transfer. Such fundamental microbiological knowledge can also help nutritional studies. Collaboration between nutritionists and microbiologists is needed to better design the research and interpret the results.
ACKNOWLEDGMENTS
This work was carried out with the support of “Cooperative Research Program for Agriculture Science & Technology Development (Project title: Investigation of rumen microbiome in Hanwoo cattle fed different diets, Project No. PJ01203103)” Rural Development Administration, Republic of Korea.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.