Stories and Challenges of Genome Wide Association Studies in Livestock — A Review
Article information
Abstract
Undoubtedly livestock is one of the major contributors to the economy of any country. The economic value of livestock includes meat, dairy products, fiber, fertilizer etc. Understanding and identifying the associations of quantitative trait loci (QTL) with the economically important traits is believed to substantially benefit the livestock industry. The past two decades have seen a flurry of interest in mapping the QTL associated with traits of economic importance on the genome. With the availability of single nucleotide polymorphism chip of various densities it is possible to identify regions, QTL and genes on the genome that explain the association and its effect on the phenotype under consideration. Remarkable advancement has been seen in genome wide association studies (GWAS) since its inception till the present day. In this review we describe the progress and challenges of GWAS in various livestock species.
INTRODUCTION
Identifying genes and quantitative trait locus (QTLs) in the genome, that are associated with any phenotype is like finding a needle in the haystack. However, identifying genes associated with traits of economic importance is not something new. In 90’s QTL mapping was largely based on microsatellite markers (Lipkin et al., 1998) whereas these days with the advent of whole genome sequencing technologies and availability of affordable whole genome single nucleotide polymorphisms (SNP) panels, SNP along with the phenotype and pedigree information are utilized for mapping. Till date hundreds of QTLs have been identified in various independent studies. The QTL database (http://www.animalgenome.org/QTLdb) maintained by United States Department of Agriculture. can be browsed to find detailed information on QTLs of interest. In the field of human genetics genome wide association studies (GWAS) had successfully identified the QTLs associated with numerous complex diseases (McCarthy et al., 2008). The first successful GWA study was published in 2005 by Klein et al. (2005) where the group carried out a genome wide scan of polymorphisms, on humans, associated with age-related macular degeneration and found two SNPs which had significantly altered allele frequency when comparing with healthy controls. In livestock GWAS has gained popularity in mapping QTL to the trait of economic importance like meat quality and quantity, sensory panel evaluation, calving ease, milk yield, fat and protein percentage, fertility traits, egg production etc., to mention a few. Usually, due to different genetic architecture of breeds and polygenic nature of the complex traits, different regions and different genes are found to be associated with the same trait in different breeds of the same species. If performed carefully, GWAS has proved to be an ideal method to identify genes associated with various phenotypes and to elucidate the mechanisms of the complex traits. In this paper we review the success stories of GWA studies (Table 1, Supplementary Tables S1 and S2) along with major challenges in the livestock species.
Genome wide association studies carried out in cattle
GENOME WIDE ASSOCIATION STUDIES IN CATTLE
With a dedicated effort of more than 300 scientists from 25 different countries in a time of 6 years, the first bos taurus (female Hereford) whole genome assembly was published in the year 2009 (Zimin et al., 2009). In the same year Matukumalli et al. (2009) described a cost-effective and efficient approach for fabricating a customized genotyping assay interrogating 54,001 single nucleotide polymorphism (SNP) loci to support GWA applications in cattle which was followed by researchers across the world. Commercial availability of the SNP genotyping assay (Illumina, San Diego, CA, USA) has expedited the GWA studies in all livestock species including cattle, pig, sheep, goat, chicken etc. There are number of GWA studies that were published after the availability of SNP chip.
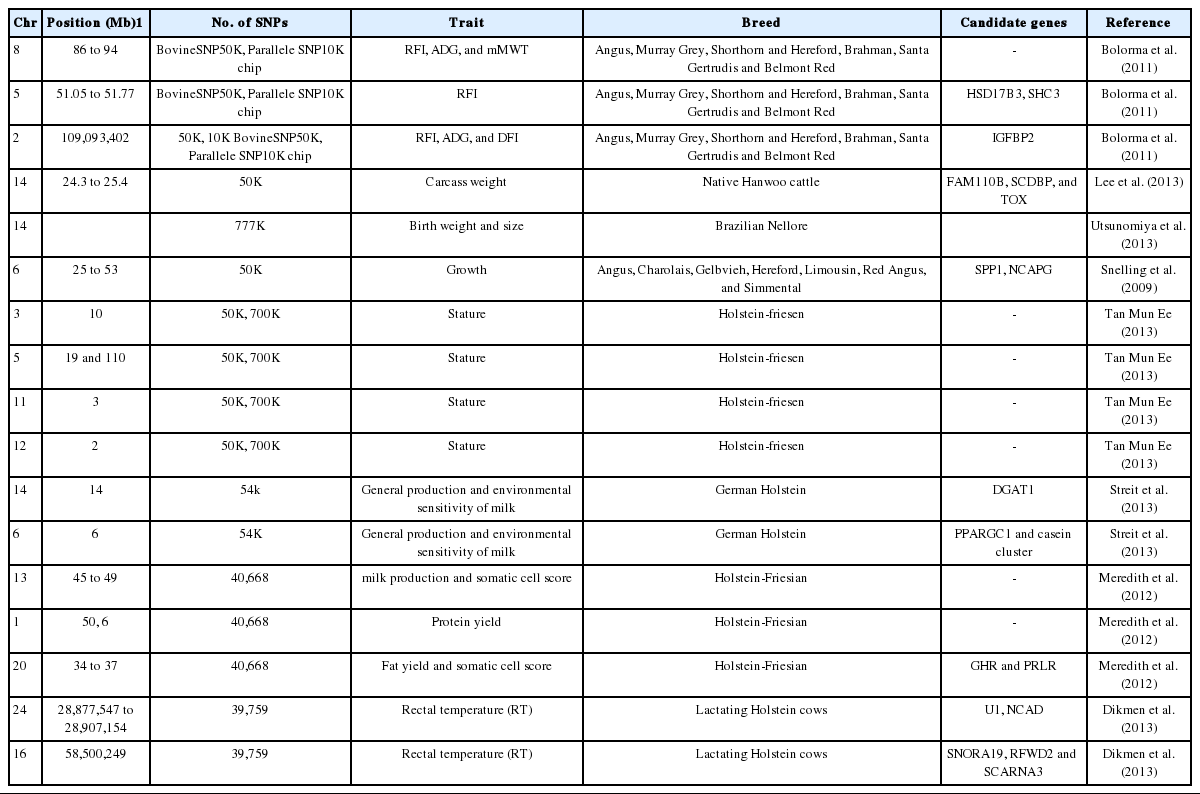
For the present study we thoroughly reviewed the available literature that reported SNPs associated with various traits in different milk and meat type livestock species. For the meat production industry it is in their favor to have big stature of the animal and better eating quality of the meat (tenderness, juiciness, taste etc.). Bolorma et al. (2011) performed a GWAS for feedlot and growth traits in 3 beef type cattle which were Bos taurus, Bos indicus and Bos taurus×Bos indicus. The 4 breeds (Angus, Murray Grey, Shorthorn, and Hereford) were Bos taurus, 1 breed (Brahman) was Bos indicus and 2 breeds (Santa Gertrudis and Belmont Red) were Bos taurus×Bos indicus synthetic breeds. This paper reported the results of a GWAS for traits related to weight, height and residual feed intake in beef and dairy cattle genotyped using 50K and 10K SNP chips, with more focus on residual feed intake (RFI). In their study they reported the association of SNPs with the RFI on chromosome 5 and 8. Bos taurus autosome 8 (86 Mbp to 94 Mb) contains several SNPs that are significant for RFI in the 50K or 10K experiments and one SNP was significant in both. The same region was found to harbour SNPs that were significantly associated with average daily gain (ADG) and midpoint metabolic weight. On BTA 5 significant associations with RFI in both the 10K and 50K datasets was found at 51.05 to 51.77 Mb region. A gene encoding hydroxysteroid (17-beta) dehydrogenase 3, which is important for steroid metabolism and another gene encoding steroid receptor coactivator (SRC) homology 2 domain containing-transforming protein C3 (SHC3) lies within this 51.05 to 51.77 Mbp region. The SRC is a signal transduction protein, involved in recognition of phosphorylated tyrosine. In humans, SHC3 play a role as a signalling adaptor that couples activated growth factor receptors to signalling pathway in neurons and is also involved in the signal transduction pathways of neurotrophin-activated Trk receptors in cortical neurons. The 3 SNP on BTA 2 situated near 109 Mb (within 42 kbp in both sides) had significant effects for the feedlot RFI, ADG, and daily feed intake traits. The gene for insulin like growth factor binding protein 2 is located on BTA2 near 109 Mb. The SNPs reported from the GWAS performed by bolorma et al. (2011) found on BTA 8, 11, 17, 18, 21, 22, 24, 25, and 26 were close to those reported by Sherman et al. (2009) and Nkrumah et al. (2007). Many SNPs on BTA 3, in a region from 102.159 Mb to 109.411 Mb were associated with different growth traits in the beef datasets as well as stature in the dairy dataset. Later in 2013 Lee et al. (2013) reported association of SNPs on BTA14 with carcass weight in Korean cattle (Hanwoo). They reported a major QTL at a region spanning 24.3 to 25.4 Mb on BTA14 and identified 6 SNPs to be significantly associated with carcass weight in this region. They found 3 genes family with sequence similarity 110, member B (FAM110 B), syndecan binding protein (SCDBP), and thymocyte selection-associated high mobility group box (TOX) that were closer to the most significant SNPs in their study. However, they did not find any association with (pleiomorphic adenoma gene 1 (PLAG1) gene, which is already known to be associated with bovine stature, in hanwoo cattle which could be attributed to i) different genetic architecture of hanwoo cattle and ii) multigene effect in which multiple genes in the same QTL region affect correlated traits in cattle. Utsunomiya et al. (2013) also reported that the QTLs on BTA14 which are associated with body size in taurine cattle (Bos primigenius taurus) also affect birth weight and size in zebu cattle (Bos primigenius indicus). They concluded it based on a GWA study, using 777K Bovine SNP chip, on Brazilian Nellore cattle. Snelling et al. (2010) performed a Genome-wide association study, using Bovine 50k SNP bead chip, for growth in crossbred beef cattle. Progeny and grandprogeny of 150 sires representing 7 breeds (Angus, Charolais, Gelbvieh, Hereford, Limousin, Red Angus, and Simmental) was genotyped for the study. Most SNP associated with direct growth were located on BTA 6. Six or more were found on BTA 7, 11, 14, and 20, and BTA 10 and 23 each had a single SNP. Most of the SNPs strongly associated with direct growth were found between 25 and 53 Mb on BTA 6. The QTL already reported for birth weight (Casas et al., 2000; Kneeland et al., 2004; Gutierrez-Gil et al., 2009), pre- and postweaning body weight gain (Yeo et al., 2003; Kneeland et al., 2004), and yearling weight (Casas et al., 2000) in beef cattle were located in this same region on BTA6. The 77 genes in the region, including secreted phosphoprotein 1 at 37.5 Mb, which is known to affect growth traits were also found in this region on BTA6. Among the most significant associations were the 5 SNPs, all in strong linkage disequilibrium, located from 38.1 to 38.3 Mb on BTA 6. This block encompasses a single annotated gene, non-SMC condensin I complex, subunit G (NCAPG), which functions in protein binding and cell division. The segment surrounding NCAPG on the mouse genome corresponds to Noq1, a QTL related to 12- and 22-wk body weight at 22-wk fat content of mice (Kluge et al., 2000). Human QTL for body mass index (Stone et al., 2002; Arya et al., 2004), fat percentage (Norman et al., 1997), and subcutaneous abdominal fat (Perusse et al., 2001) are within the region of homo sapiens autosome 4 which is syntenic to this segment of BTA 6. Tan Mun Ee (2013) carried GWAS for stature in New Zealand dairy cattle. The dairy cattle included Holstein-friesen, Jersey, and Holstein-friesen×Jersey crossbreed bulls. The study identified BTA 2, 3, 4, 5, 6, 11, 12, 14, 24 in Holstein-friesen, BTA 9,10,12,18,19,25 in jersey and BTA 1, 3, 4, 5, 7, 9, 10, 14, 18, 22, and 24 in Holstein-friesen μ×Jersey to be significantly associated with stature.
Streit et al. (2013) also identified BTA14 as one of the major chromosomes harboring SNPs associated with protein yield, fat yield, and milk yield in daughter of German Holstein sires. The association might be due to the effect of diacylglycerol O-acyltransferase 1 (DGAT 1) gene which is known to segregate and affect all milk traits in this population. Also BTA6 was identified to affect the general production of all three milk traits. Peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PPARGC1A) and casein cluster is known to reside on chromosome 6 in cattle and affect the fat yield, protein yield and protein percentage. BTA5 and BTA 20 were also identified to harbor the SNPs affecting the milk traits. In the Norwegian Red, milk production QTL for Environmental Sensitivity was identified on BTA2, BTA6, BTA7, and BTA16 (Lillehammer et al., 2007, 2008). Meredith et al. (2012) carried out a GWA study in Holstein-Friesian cattle of Ireland for milk production and somatic cell score. In their study using a single marker regression approach they identified 276 novel SNPs in sires that were associated to milk production and somatic cell score. There were 103 associations in common between the sires and cows across all the traits. They found a region from ~45 to 49 Mb on chromosome 13 to be highly associated with milk yield in the population of Holstein-Friesian sires. For protein yield the most significantly associated SNP across all chromosomes was rs42327956 located at ~50, 6 Mb on chromosome 1. For somatic cell score most significant association was detected on chromosome 20, however there were other associations detected at chromosome 6, 10, and 15 too. Significant novel associations were detected on chromosome 20 for fat yield and somatic cell score close to the growth hormone receptor (GHR) and prolactin receptor (PRLR) genes reported to be associated with milk production traits and somatic cell score. Any kind of stress in livestock species can considerably affect their production traits. Dikmen et al. (2013) performed a GWAS for rectal temperature (RT) during heat stress, which is known to affect production, fertility, and health of dairy cattle, in lactating Holstein cows and the largest proportion of SNP variance (0.07% to 0.44%) was explained by markers flanking the region between 28,877,547 and 28,907,154 bp on BTA 24. That region is flanked by U1 (28,822,883 to 28,823,043) and cadherin-2 (NCAD) (28,992,666 to 29,241,119). In addition, the SNP at 58,500,249 bp on BTA 16 explained 0.08% and 0.11% of the SNP variance for 2- and 3-SNP analyses, respectively. That contig includes small nucleolar RNA, H/ACA box 19 (SNORA19), ring finger and WD repeat domain 2 (RFWD2), and small Cajal body-specific RNA 3 (SCARNA3). Other SNPs associated with RT were located on BTA 16 (close to centrosomal protein 170 kDa [CEP170] and phospholipase D family, member 5 [PLD5]), BTA 5 (near solute carrier organic anion transporter family, member 1C1 [SLCO1C1] and phosphodiesterase 3A [PDE3A]), BTA 4 (near kelch repeat and BTB (POZ) domain containing 2 [KBTBD2]) and LSM5 [LSM5 homolog]), and BTA 26 (located in glutamic-oxaloacetic transaminase 1 [GOT1], a gene implicated in protection from cellular stress). Genes namely ATP-binding cassette, sub-family A (ABC1), member 12 (ABCA12), fibronectin leucine rich transmembrane protein 2 (FLRT2), LIM homeobox 4 (LHX4), mitogen-activated protein kinase kinase kinase 5 (MAP3K5), nutritionally-regulated adipose and cardiac-enriched (NRAC), netrin G1 (NTNG1), phosphatidylinositol glycan anchor biosynthesis, class N (PIGN), and zinc finger protein 75a (ZNF75A) were identified to be associated with calf birth weight in Holstein cattle. A pathway that includes the rho-associated, coiled-coil containing protein kinase (ROCK) gene, which is involved in placental function in the human, as well as other developmental genes (e.g., family kinase-interacting [FAK] and serine/threonine-protein kinase [PAK]) for calf birth weight in Holstein cattle was also identified using single nucleotide polymorphisms (Cole et al., 2013). Murdoch et al. identified several regions of the genome that were significantly associated with the incidence of bovine spongiform encephalopathy in European Holstein cattle on chromosomes 2, 14, 16, 20, 21, and 28. A GWAS was conducted to identify four loci associated with conception rate in Holsteins, a breed known as the world’s most productive dairy cattle. These loci harbored two gap junction-related genes, plakophilin 2 (PKP2) and cortactin-binding protein 2 Nterminal like (CTTNBP2NL), and two neuroendocrine-related genes, SET domain containing 6 (SETD6) and calcium channel, voltage-dependent, beta 2 subunit (CACNB2). The GWAS uncovered the unexpected roles for these genes and provided a potential solution for the problem of declining conception rates in the livestock industry (Sugimoto et al., 2013).
In a total of 461 animals and 40,657 SNPs a GWAS identified genomic regions for fatty acid composition in Japanese Black cattle (Ishii et al., 2013). They used genome-wide rapid association using mixed model and regression (GRAMMAR) and genomic control approaches to estimate the associations between genotypes and fatty acid composition. In addition, two SNPs in fatty acid synthase (FASN) (T1952A) and stearoyl-CoA desaturase (SCD) (V293A) genes were also genotyped. Association analysis revealed that 30 significant SNPs for several fatty acids (C14:0, C14:1, C16:1, and C18:1) were located on the BTA19. The FASN gene was found within this region but the FASN mutation had no significant effect on any traits. They also detected one significant SNP for C18:1 on BTA23 and two SNPs for C16:0 on BTA25. The region around 17 Mb on BTA26 harbored two significant SNPs for C14:1 and a SNP in SCD gene in this region showed the strongest association with C14:1. This study demonstrated novel candidate regions in BTA19, 23 and 25 for fatty acid composition.
In Norwegian Red cattle Olsen et al. (2011) identified quantitative trait loci for fertility and milk production on BTA12. The most interesting result was found for non-return rate for heifers on BTA12, at a position where significant associations with several milk production traits have previously been found. Subsequent fine-mapping verified the presence of a QTL at 18 Mb with opposite effects on non-return rate and milk production. None of the other reproduction QTL were found to affect milk production, and these are therefore of considerable interest for use in marker-assisted selection.
GENOME WIDE ASSOCIATION STUDIES IN PIGS
The pig genome was sequenced and characterised by the Swine Genome Sequencing Consortium formed in 2003 (Schook et al., 2005). Groenen et al. (2012) published the first high-quality draft of pig genome sequence. With the availability of PorcineSNP chip it became possible to carry out whole-genome association studies, determine the genetic merit, identify quantitative trait loci, and carry out comparative genetic studies. Duijvesteijn et al. (2010), based on the Illumina Porcine 60K+SNP Beadchip, revealed several areas of the genome responsible for variation of androstenone levels in intact boars. Nine hundred eighty-seven pigs divergent for androstenone concentration from a commercial Duroc-based sire line were genotyped for their study. The association analysis revealed that androstenone levels in fat tissue were significantly affected by 37 SNPs on pig chromosomes sus scrofa chromosome1 (SSC1) and SSC6. Among them, the 5 most significant SNPs explained together 13.7% of the genetic variance in androstenone. On SSC6, a larger region of 10 Mb was shown to be associated with androstenone covering several candidate genes potentially involved in the synthesis and metabolism of androgens. Besides known candidate genes, such as cytochrome P450 A19 (CYP2A19), sulfotransferases sulfotransferase family, cytosolic, 2A (SULT2A1), dehydroepiandrosterone (DHEA)-preferring, member 1, and sulfotransferase family, cytosolic, 2B, member 1 (SULT2B1), also new members of the cytochrome P450 CYP2 gene subfamilies and of the hydroxysteroid-dehydrogenases were found. In addition, the gene encoding the s-chain of the luteinizing hormone (LHB) which induces steroid synthesis in the Leydig cells of the testis at onset of puberty maps to this area on SSC6. Interestingly, the gene encoding the α-chain of LH is also located in one of the highly significant areas on SSC1. Major genetic factors on SSC1 and SSC6 showing moderate to large effects on androstenone concentration were identified in this commercial breeding line of pigs.
Genome-wide association analysis identified single QTL for the estimated breeding values (EBVs) pH one hour post mortem (pH1) and carcass length were on pig chromosome (SSC) 14 and SSC 2, respectively in Swiss Large White breed. Two QTL for the EBV rear view hind legs were on SSC 10 and SSC 16 Becker et al. (2013).
Eating behaviour of pigs was studied by Do et al. (2013). One thousand and one hundred thirty boars were genotyped using the Illumina Porcine SNP60 BeadChip. Musashi RNA-binding protein 2 (MSI2) gene on chromosome (SSC) 14 was very strongly associated with NVD (number of daily visits to feeder). Thirty-six SNPs were located in genome regions where QTLs have previously been reported for behavior and/or feed intake traits in pigs. The regions: 64 to 65 Mb on SSC 1, 124 to 130 Mb on SSC 8, 63 to 68 Mb on SSC 11, 32 to 39 Mb, and 59 to 60 Mb on SSC 12 harbored several significant SNPs. Synapse genes (gamma-aminobutyric acid (GABA) A receptor, rho 2 [GABRR2], protein phosphatase 1, regulatory subunit 9B [PPP1R9B], synaptotagmin I [SYT1], gamma-aminobutyric acid (GABA) A receptor, rho 1 [GABRR1], Ca++-dependent secretion activator 2 [CADPS2], discs, large homolog-associated protein 2 [DLGAP2], and golgi-associated PDZ and coiled-coil motif containing [GOPC]), dephosphorylation genes (protein phosphatase, Mg2+/Mn2+ dependent, 1E [PPM1E], dual adaptor of phosphotyrosine and 3-phosphoinositides [DAPP1], protein tyrosine phosphatase, non-receptor type 18 [PTPN18], protein tyrosine phosphatase, receptor-type, Z polypeptide 1 [PTPRZ1], protein tyrosine phosphatase, non-receptor type 4 [PTPN4], myotubularin related protein 4 [MTMR4], and RNA guanylyltransferase and 5′-phosphatase [RNGTT]) and positive regulation of peptide secretion genes ( growth hormone releasing hormone [GHRH], neuronatin [NNAT], and transcription factor 7-like 2 [TCF7L2]) were highly significantly associated with feeding behavior traits.
Schneider et al. (2012) performed the genome-wide association study of swine farrowing traits and identified 124 statistically significant SNPs. Traits in the study included total number born (TNB), number born alive (NBA), number born dead (NBD), number stillborn (NSB), number of mummies (MUM), total litter birth weight (LBW), and average birth weight (ABW). Eleven QTL were found for TNB, 3 on SSC1, 3 on SSC4, 1 on SSC13, 1 on SSC14, 2 on SSC15, and 1 on SSC17. For NBA 14 QTL, 4 on SSC1, 1 on SSC4, 1 on SSC6, 1 on SSC10, 1on SSC13, 3 on SSC15, and 3 on SSC17 were identified. A single NBD QTL was found on SSC11. No QTL were identified for NSB or MUM. Thirty-three QTL were found for LBW, 3 on SSC1, 1 on SSC2, 1 on SSC3, 5 on SSC4, 2 on SSC5, 5 on SSC6, 3 on SSC7, 2 on SSC9, 1 on SSC10, 2 on SSC14, 6 on SSC15, and 2 on SSC17. A total of 65 QTL were found for ABW, 9 on SSC1, 3 on SSC2, 9 on SSC5, 5 on SSC6, 1 on SSC7, 2 on SSC8, 2 on SSC9, 3 on SSC10, 1 on SSC11, 3 on SSC12, 2 on SSC13, 8 on SSC14, 8 on SSC15, 1 on SSC17, and 8 on SSC18. Several candidate genes have been identified that overlap QTL locations among TNB, NBA, NBD, and ABW. These QTL when combined with information on genes found in the same regions could provide useful information that could be exploited for marker assisted selection, or genomic selection and for better management practices in commercial pig populations. Hematological traits, which are important indicators of immune function in animals, have been commonly examined as biomarkers of disease and disease severity in humans and animals. Genome-wide significant QTL provide important information for use in breeding programs of livestock species. Luo et al. (2012) carried out a genome-wide association study of porcine hematological parameters in a large white×minzhu F2 resource population.In this study seven hematological parameters were measured hematocrit (HCT), hemoglobin, mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration, mean corpuscular volume (MCV), red blood cell count (RBC) and red blood cell volume distribution width (RDW) were measured. Data was analyzed using genome-wide rapid association using the mixed model and regression-genomic control (GRAMMAR-GC) method and a total of 62 genome-wide significant and 3 chromosome-wide significant SNPs were detected to be associated with hematological parameters. For hematological traits significant association was found on SS7 in the region spanning from 34.6 to 36.5 Mb. 7 associations for HCT and 5 associations for hepatocyte growth factor were found. Four SNPs within the region of 43.7 to 47.0 Mb and fifty-five SNPs within the region of 42.2 to 73.8 Mb on SSC8 showed significant association with MCH and MCV, respectively. At chromosome-wide significant level, one SNP at 29.2 Mb on SSC1 and two SNPs within the region of 26.0 to 26.2 Mb were found to be significantly associated with RBC and RDW, respectively. Many of the SNPs were located within previously reported QTL regions and appeared to narrow down the regions compared with previously described QTL intervals. A total of seven significant SNPs were found within six candidate genes signal peptide, CUB domain, EGF-like 3 (SCUBE3), kinase insert domain receptor (KDR), tryptophan 2,3-dioxygenase (TDO), insulin-like growth factor binding protein 7 (IGFBP7), ADAM metallopeptidase with thrombospondin type 1 motif, 3 (ADAMTS3), and alpha-fetoprotein (AFP). In addition, the v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) gene, which has been previously reported to relate to hematological parameters, was located within the region significantly associated with MCH and MCV and could be a candidate gene. These results of this study may lead to a better understanding of the molecular mechanisms of hematological parameters in pigs.
CHICKEN
Body weight and Growth are two of the most important economic traits for the poultry industry. So it is desirable to identify the DNA polymorphisms affecting these traits. Various GWAS studies have found Gallus gallus chromosomes 1 and 4 (GGA1, GGA4) to be of particular interest as far as the growth traits are concerned. Gu et al. (2011) with the use of 60 k SNP panel in a chicken F2 resource population of china agricultural university, cross between Silky Fowl and White Plymouth Rock reported a total of 26 SNP effects involving 9 different SNP markers on chicken chromosome 4 (GGA4). A region ~8.6 Mb in length (71.6 to 80.2 Mb) was identified to have a large number of significant SNP effects for late growth during weeks 7 to 12. The LIM domain-binding factor 2 (LDB2) gene in this region had the strongest association with body weight for weeks 7 to 12 and with ADG for weeks 6 to 12. This GGA4 region was previously reported to contain body weight QTL. Also, Johansson et al. (2010) utilized 60 K SNP chip and reported a region spanning 60 to 80 Mb on GGA4 to be under recent and ongoing selection in chicken lines with divergent selection on body weight for up to 50 generations. Gu et al. (2011) also found three significant SNP effects on GGA1 and GGA18. The A allele of GGaluGA266058 in the LDB2 gene was found to have the strongest association with late growth (7 to 12 weeks). A polymorphism (GGA_rs16432721) positioned 92 kb downstream of the TBC1 (tre-2/USP6, BUB2, cdc16) domain family, member 1 (TBC1D1) gene was highly significant for body weight at 12 weeks of age. Many other SNPs near LOC769270 gene had strong association with late growth. One SNP on GGA1 in the oculocutaneous albinism II (OCA2) gene had highly significant effects on body weight in weeks 11 to 12. For early growth traits, only one SNP (GGaluGA118136) on GGA18 had significant association with body weight at 2 weeks of age. Various genes like glypican 6 (GPC6) gene, GPC5 gene, and gga-mir-17–92 cluster, Popeye domain-containing protein 1, Opioid-binding protein/cell adhesion molecule-like, Cbfa2t2 were found to be associated with body weight at different growth stages.
In Beijing-You chickens, a region on chromosome 4 (GGA4) in 1.06 Mb region (78. 4 to 79.5 Mb), including seven significant SNPs and four candidate genes ligand dependent nuclear receptor corepressor-like (LCORL), leucine aminopeptidase 3 (LAP3), LDB2, transmembrane anterior posterior transformation 1 (TAPT1), were found to have association with carcass weight and eviscerated weight (Liu et al., 2013). The possibility of gap junction protein alpha 1 gene as a functional gene was also reported. Five SNPs of significance were identified, located on GGA2 and GGA27. Three significant SNPs on GGA2 were found to associate with dry matter content in breast and were located in the vicinity of or within angiopoietin 1 (ANGPT1). Intramuscular fat in breast (IMFBr). Two significant SNPs were located on GGA2 and GGA5 identified. The SNP on GGA2 was 120.9 kb away from cholecystokinin. The SNP on GGA5 was 159.7 kb from toll interacting protein. Six significant SNPs associated with dry matter content in thigh were identified on GGA1, GGA2, and GGA8. The SNP on GGA2 was found 208.4 kb away from suppressor of cytokine signaling 6 (SOCS6). The SNP on GGA8 was 8.5 kb from UMP-CMP kinase (CMPK1).The four SNPs on GGA1 clustered within a 1.30 Mb region with no annotated genes nearby. Eleven significant SNPs were identified on GGA2, GGA4, and GGA7. The eight SNPs on GGA2 were distributed within a 2.06 Mb region and only one gene (Polypeptide N-acetylgalactosaminyltransferase 1, GALNT1) was identified in the vicinity. Two SNPs on GGA4 located within protocadherin 19 or 286.3 kb away from diaphanous homolog 1. The SNP on GGA7 was identified 143.9 kb from the secreted phosphoprotein 2. In the same Chinese chicken breed Beijing-You and a commercial broiler line (Cobb-Vantress) 33 association signals for 10 meat quality traits were detected and 14 candidate genes were identified. A total of 14 genes associated with IMFBr, meat color, abdominal fat weight (AbFW), and abdominal fat percentage (AbFP), were differentially expressed between the high and low phenotypic groups. These genes are, therefore, prospective candidate genes for meat quality traits: protein tyrosine kinase and microsomal glutathione S-transferase 1 for IMFBr; collagen, type I, alpha 2 for meat color and RET proto-oncogene (RET), natriuretic peptide B and sterol regulatory element binding transcription factor 1 for the abdominal fat (AbF) traits (Sun et al., 2013).
Number of eggs and age at first egg are two desirable production traits in layer type chicken breeds. A SNP associated with egg number was found to be located in the intron12 of growth factor receptor-bound protein 14. (GRB14) gene that encodes a growth factor receptor-binding protein. In human and mammals, GRB14 mRNA was found to be expressed at high level in the ovary, liver, kidney, skeletal muscle and so on although the function of GRB14 in chicken is undefined, it may combine with the insulin-like growth factor system to influence egg production in layers. A SNP in the intron2 of odd Oz/ten-m homolog 2 (ODZ2) gene was revealed to be associated significantly with Age at first egg. The ODZ2, also known as Teneurin-2, encodes a neuronal cell surface protein and plays an important role in development of nervous system. It was found that Teneurin are expressed prominently in developing chicken brain, and especially in the visual system including retina and optic tectum. Teneurin-2 may have effect on the sexual maturity of chickens (Liu, 2011).
The GWA studies have provided an insight into the genetics of Marek’s disease and Newcastle disease. Marek’s disease, named after a Hungarian veterinarian, Jozsef Marek, is a highly contagious viral disease caused by herpesvirus. Results from a study by Wolc et al. (2012) suggested some regions on chromosomes GGA 2, 3, 4, 9, 15, 18, and 21 to be associated with Marek’s disease resistance. Many genes, such as serpins, BCL-2 proteins; tumor necrosis factor receptor superfamily, member 11a (TNFRSF11A), unc-13 homolog D (UNC13D), sphingosine, ArfGAP with FG repeats 1 (AGFG1), mitogen-activated protein kinase kinase 4 (MAP2K4), IGFBP7; receptor (TNFRSF)-interacting serine-threonine kinase 1 (RIPK1), were identified. Also, the region at about 100 Mb from the proximal end of chicken chromosome 1, including the roundabout, axon guidance receptor, homolog 1 (ROBO1) and ROBO2 genes, has a strong effect on the antibody response to the Newcastle disease virus (NDV) in chickens. This study paves the way for further research on the host immune response to NDV (Luo et al., 2013).
LIMITATIONS AND CHALLENGES OF GENOME WIDE ASSOCIATION STUDIES
Along with the success stories there are quite a few unsuccessful ones too. As the cost of genotyping has fallen down considerably, it is now in the reach of even the small laboratories with no big funding. Also with the ample information and tutorials available on the web, analysis remains not such a big question. So in zest of performing the analysis one might end up reporting false associations. So it becomes necessary to meticulously design the study and perform it. Quality control of the data is one of the most important steps to minimize the errors in a GWA study. The approaches presented so far rely on two fundamental assumptions: first, the population under study must be genetically homogeneous, i.e. there should be no population stratification; second, all subjects in the samples must represent statistically independent units drawn from that population. If not taken care of, the tests of association may lead to spurious associations or may have inflated type I error rates. Another scenario is that the related individuals share both causal and non-causal alleles, and that linkage disequilibrium between these sites can lead to artifacts. A powerful method to deal with the artifacts was first developed in the field of animal breeding: mixed models that handle population structure by accounting for the amount of phenotypic covariance. Mixed models have been applied to GWAS, and can markedly reduce the number of false positive associations. Lack of statistical knowledge remains one of the major glitches in GWA projects. Only if performed carefully could GWAS lead to meaningful and valuable results.
CONCLUSION
Genetics has come a long way since 1983 (Soller and Beckman 1983; Beckman and Soller 1983), when the genetic markers were first used for the improvement of crops and livestock. The advancement in technology go hand in hand with genetics and genomics. With ever advancing technology and better knowledge of genetic mechanisms, we are surely a step closer to the understanding of complex traits. The challenges of gwas include carefully and strategically choosing a homogeneous population for the study and to account for population stratification. The statistical models, if carefully chosen, can be useful to minimize the chances of false associations.
Supplementary Data
ACKNOWLEDGMENTS
This study was supported by awards from the AGENDA project (Grant no. PJ006405) and Molecular Breeding program (PJ0081882014) of Next Generation BIOGREEN21 project in the National Institute of Animal Science, RDA, Korea.