A Genome-wide Scan for Selective Sweeps in Racing Horses
Article information
Abstract
Using next-generation sequencing, we conducted a genome-wide scan of selective sweeps associated with selection toward genetic improvement in Thoroughbreds. We investigated potential phenotypic consequence of putative candidate loci by candidate gene association mapping for the finishing time in 240 Thoroughbred horses. We found a significant association with the trait for Ral GApase alpha 2 (RALGAP2) that regulates a variety of cellular processes of signal trafficking. Neighboring genes around RALGAP2 included insulinoma-associated 1 (INSM1), pallid (PLDN), and Ras and Rab interactor 2 (RIN2) genes have similar roles in signal trafficking, suggesting that a co-evolving gene cluster located on the chromosome 22 is under strong artificial selection in racehorses.
INTRODUCTION
The Thoroughbred is best known as a racehorse breed. They have been bred exclusively for structural and functional adaptations that contribute to athletic performance phenotypes (Williamson and Beilharz, 1998) since their establishment about 300 to 400 years ago (Cunningham et al., 2001; Hill et al., 2002). Various traits, such as racing time, rank, annual earnings, and others, are used to measure a horse’s racing performance (Ricard, 1998). Previous genome-wide association studies (GWAS) have identified a handful of genetic markers in the myostatin (MSTN) (Grobet et al., 2003) with racing performance (Binns et al., 2010; Hill et al., 2010; Tozaki et al., 2010). In dogs, mutations in MSTN were found to be associated with athletic performance (Tozaki et al., 2010; McGivney et al., 2012). A scan for positive selection based on a microsatellite data detected hundreds of domesticated genes in horse (Gu et al., 2009), showing that MSTN is not the only determinant of athletic prowess (Lee, 2007). Recently, comprehensive analysis of equine breed diversity suggested that genomic footprints of selection are important candidates of GWAS (Petersen et al., 2013; Park et al., 2014).
Several association studies followed by selective sweep mapping found genetic markers associated with the complex traits in dog (Vaysse et al., 2011) and cattle (Qanbari et al., 2014). Motivated by these approaches, we aimed to use selective sweeps to detect markers associated with artificial selection for racing performance in Thoroughbreds. To this end, we compared genetic diversity between Thoroughbreds and Jeju domestic breed. Jeju breed is a close relative of the ancestral horse population (Nam, 1969; Kim et al., 1999; Jansen et al., 2002). We conducted a genome-wide scan to detect putative candidate loci showing high genetic differentiation between populations as well as low genetic diversity in Thoroughbreds. On obtaining candidate loci under directional selection, we evaluated their potential effects by association study of finishing time in a large sample of Thoroughbreds. We found a large linkage-disequilibrium (LD) block of gene cluster on Equus caballus chromosome 22 (ECA22), mainly consisting of signal regulation-related genes, that is subject to artificial selection in the Thoroughbred.
MATERIALS AND METHODS
Ethics statement
Blood samples were taken from horses and ponies by trained veterinarians according to relevant international guidelines as well as national guidelines and under permission from the Korean Racing Authority and from the Jeju Provincial Livestock Institute, Korea.
Sample preparation and resequencing horse genomes
Sets of whole-blood samples were collected from 14 Thoroughbred race stallions from the Korean Racing Authority and 2 male Jeju domestic ponies (Equus caballus), Korea’s National Treasure Number 347, from the Jeju Provincial Livestock Institute, Korea. Blood (10 mL) was drawn from the carotid artery and was treated with heparin to prevent clotting.
A genomic DNA quality check (DNA QC) was carried out using an agarose gel+fluorescence-based quantification check with a standard electrophoresis using a 0.6% agarose gel and 200 ng pulse-field gel loading. Briefly, mate-pair library construction (500 bp fragment), amplification and paired-end sequencing using Illumina HiSeq system (San Diego, CA, USA) was done by National Instrumentation Center for Environmental Management (NICEM), Seoul, Korea; the steps including purifying genomic DNA, generating fragments of less than 800 bp, using blunt-ended fragments with 5′-phosphorylated ends and 3′-dA overhang, ligating adapter-modified ends, purifying the ligation product, and producing the genomic DNA library, and then, sequence data were generated using the Illumina HiSeq system (San Diego, CA, USA). The data set of 14 Thoroughbred racing stallions and 2 ‘Jeju’ lineage have been uploaded to the Sequence Read Archive (SRA) in NCBI (accession number: SRA053569).
Genotype calling from next-generation sequencing data
Pair-end sequence reads were mapped to the reference horse genome (equCab2) using the Burrows-Wheeler Aligner (BWA; version 0.6.1) with the default settings (Li and Durbin, 2009). Three open-source packages were used for downstream processing and variant calling: Picard tools, SAMtools (Anisimova and Kosiol, 2009), and Genome Analysis Toolkit (McKenna et al., 2010). Substitution calls were made with GATK UnifiedGenotyper. All calls with a Phred-scaled quality score of less than 20 were filtered out. We identified alternative homozygous and heterozygous sites in respect to the reference genome in each sample variants. Then, we used BEAGLE to infer haplotype phase and impute missing alleles for the entire set of Thoroughbred genomes simultaneously (Browning and Browning, 2007).
Data analysis
For a limited number of samples available, especially for Jeju horse, a large number of variants can substitute for sample size when estimating FST (Willing et al., 2012). Horse genomes were divided into a large window of 50 kb that produced 47,308 bins covering ~87.6% of the whole genome. An unbiased estimation of nucleotide diversity (Nei, 1987) and population differentiation (Nei and Li, 1979) based on the pairwise difference between haplotypes was calculated for each bin by using Arlequin 3.5 (Excoffier and Lischer, 2010). The ratio of nucleotide diversity of the Jeju breed to that of the Thoroughbred was standardized to have a mean of 0 and standard deviation of 1. We adopted an empirical outlier approach to identify potential targets of directional selection by obtaining top 0.1% of highly divergent regions as well as top 0.1% of strong reduction of genetic diversity in Thoroughbreds (Excoffier et al., 2009).
Validation of associated loci on Equus caballus chromosome 22
A group of 240 Thoroughbreds that were registered in the Korea Racing Authority (KRA) for breeding as stallions was genotyped for validating selective sweeps using EquineSNP50 BeadChips (Illumina, San Diego, CA, USA) in the NICEM at Seoul National University. After the quality control steps, removing single nucleotide polymorphisms (SNPs) with a percent of missing allele >5%, minor allele frequency <5%, and Hardy-Weinberg equilibrium test <10−3, 940 SNPs on ECA22 were obtained for association analysis that was conducted using linear regression in PLINK (Purcell et al., 2007). We employed a genomic control to correct for spurious association due to population stratification. The data set is available upon request.
Total records of 262,326 racing times between 1994 and 2011 in racecourses (Seoul Racecourse, Busan-Gyeongnam Racecourse) of the KRA were obtained. Each sample was assigned an estimated breeding value (EBV), which was defined as a statistical numerical prediction of the relative genetic breeding value and used to rank breeding stock for selection. When a horse has multiple records, the EBV can be used to combine the measurements in all records into one numerical value. To improve the accuracy of the EBV, we simplified the animal model by reducing unnecessary effects (racing year, racing type, and type of weight carried). The repeated-records animal model used to estimate the genetic parameters (breeding value):
where Y = the vector of observations, b = the vector of fixed effects, v = the vector of random effects, pe = the vector of permanent environmental effect: common environment, a = the vector of individual additive genetic effect, and e = the vector of residual error. X, W1, W2, and Z are design matrices for b, v, pe, and a, respectively. All parameters were estimated using the ASREML program (Gilmour et al., 2009) facilitated by the derivative-free restricted maximum likelihood method for a single-trait animal model. Using this model, we calculated the EBV for the phenotype used in this association study.
To minimize “over-correction” problem, we defined LD blocks from R2>0.7 that resulted n = 672, in total number of blocks+inter-block SNPs. We used 1/n as a cutoff for significance that has less violation of the assumption of independence (Duggal et al., 2008).
RESULTS
Sequence variations in horses
Overall, 228,696,430 sequencing paired-end reads of length 75 bp, totaling 22 Gbp, were generated for each sample. By applying a genotype-calling pipeline (see Methods for details) to them, the next-generation sequencing yielded an average of 2,586,724 variable sites per sample with a 17.1-fold read depth per base. A total of 2,709,922 SNPs segregating in Thoroughbred and Jeju breed were identified after imputation. These SNPs overlapped with 67.74% of SNPs on the EquineSNP50 BeadChips. Nucleotide diversity (π) in Thoroughbred and Jeju breed was similar across the genome. However, the mean value of nucleotide diversity (π) of the Jeju breed was slightly higher (10%) than that of the Thoroughbred.
Signatures of selection associated with racehorse domestication
High FST is indicative of diversifying selection. The low ratio of nucleotide diversity in Jeju breed to that in Thoroughbreds (πJeju/πTb) results from reduction of diversity by strong artificial selection in the Thoroughbreds. For their complex demographic history (Orlando et al., 2013), we used the outlier approach to identify candidate loci showing low π score as well as high population differentiation (FST) (Figure 1). We obtained twelve outliers as putative signatures of directional selection in the Thoroughbreds (Table 1).
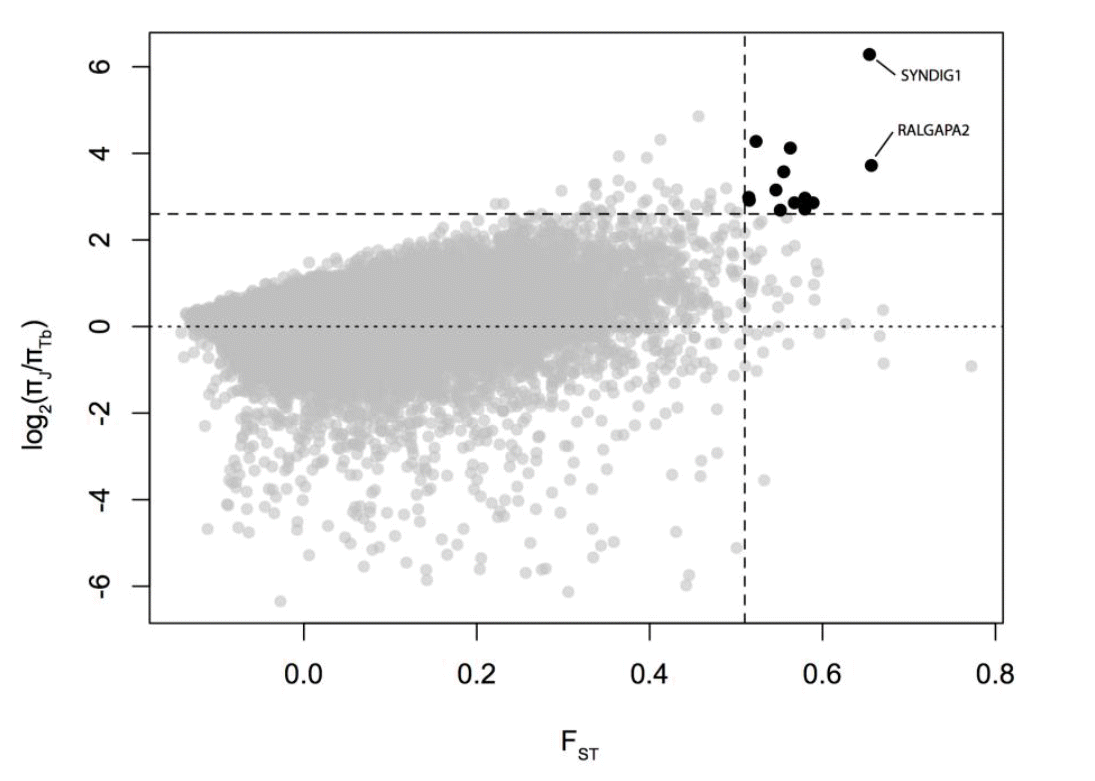
Putative signatures of selective sweeps in the Thoroughbred population. Black dots depict potential candidates of top 0.1% (vertical dashed line) of FST as well as top 0.1% (horizontal dashed line) of the ratio of π in Thoroughbreds to that in Jeju breed. Dotted line depicts π in Thoroughbred being equal to that in Jeju breed.

Potential domesticated genes associated with strong signatures in racing horse population
Two loci on ECA22 showing strongest selection signals were involved in actin-based cell motility and synapse contents. One of them is Ral GApase alpha 2 (RALGAPA2; Figure 2A), showing a clear pattern of reduced nucleotide sequence diversity up- and down-stream of focal sites. The RALGAPA2 produces multifunctional proteins that regulate a variety of cellular processes, including cell signal trafficking (Thomas et al., 2003). Another region showing a peak of FST value was located in a 3′ end of synapse differentiation induced gene 1 (SynDIG1, Figure 2B). The SynDIG1 encodes a conserved type II transmembrane protein that may play an essential role in the central regulation of excitatory synaptic strength (Kalashnikova et al., 2010). This region overlapped with one of Thoroughbred-specific putative signatures of selection that have been identified previously in large-scale population genetics study using SNP arrays (Petersen et al., 2013).
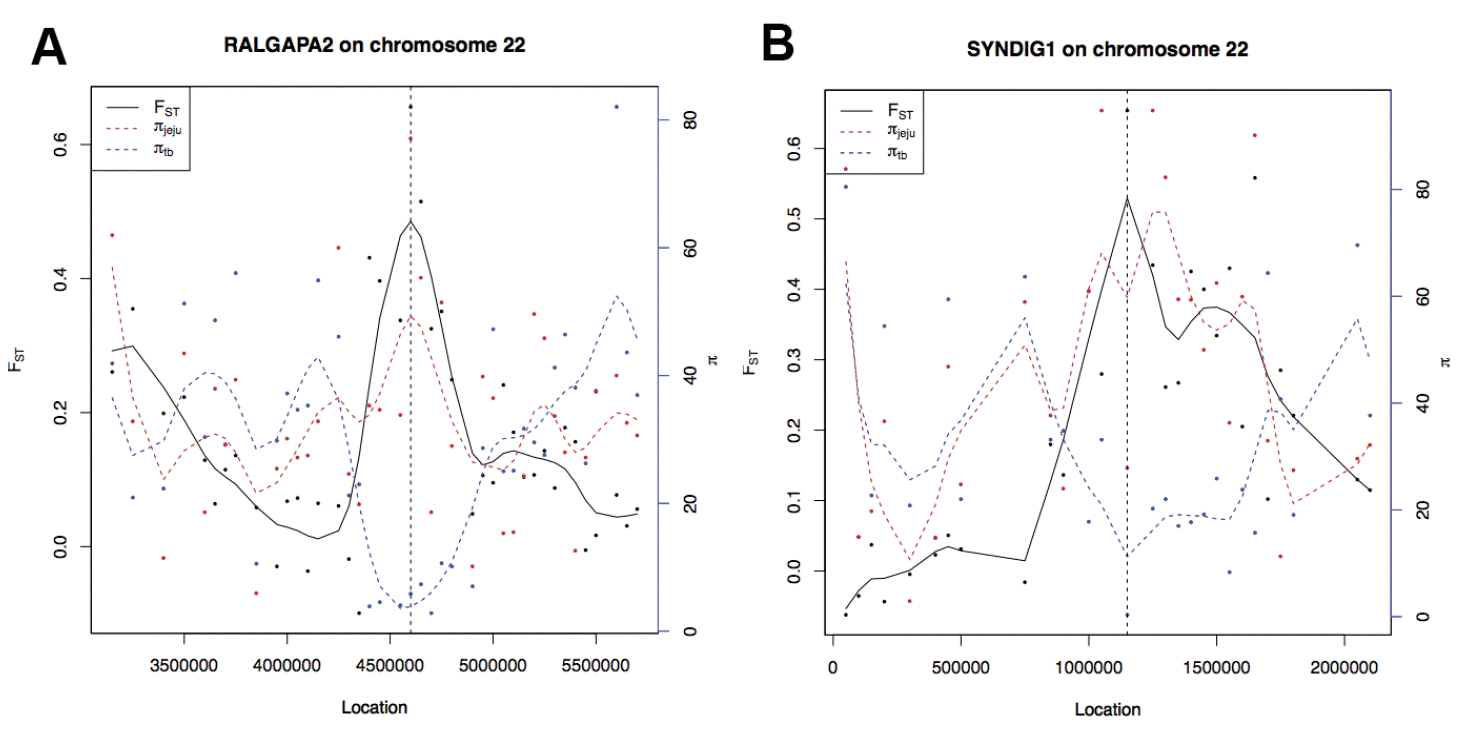
Genome regions with strongest signatures of selective sweep. The vertical line depicts the focal site proximal to RALGAPA2 (A) and SYNDIG1 (B), where the FST value (black line) is high and the π in the Thoroughbred (blue dashed line) is relatively lower than that in Jeju (red dashed line) in regions. RALGAPA2, Ral GApase alpha 2; SYNDIG1, synapse differentiation induced gene 1.
Intense artificial selection contributing to racing performance on chromosome 22
We further investigated the role of those candidates on ECA22 by conducting an association study to evaluate association of SNPs within them to racing performance in Thoroughbred. We estimated effects of SNPs on EBV of the finishing time for independent groups of 240 Thoroughbreds. With a significance threshold after multiple tests being 0.0015, the identified significant SNPs were significantly associated with racing performance. Half of them were positioned in genes (Table 2), including RALGAPA2. Furthermore, we found a long LD block at RALGAPA2 (Figure 3), reflecting strong selection has been acting on the region to have high level of haplotype homozygosity. However, the other half distributed in intergenic regions, suggesting that, even though significant SNPs identified by association studies can be used as genetic markers for breeding value prediction and breeding purpose, it may be difficult to link these markers to the genetic basis underlying the quantitative traits. Thus, there results provide strong evidence that RALGAPA2 plays critical role in improving racing performance in horses.

Significant SNPs associated with racing performance
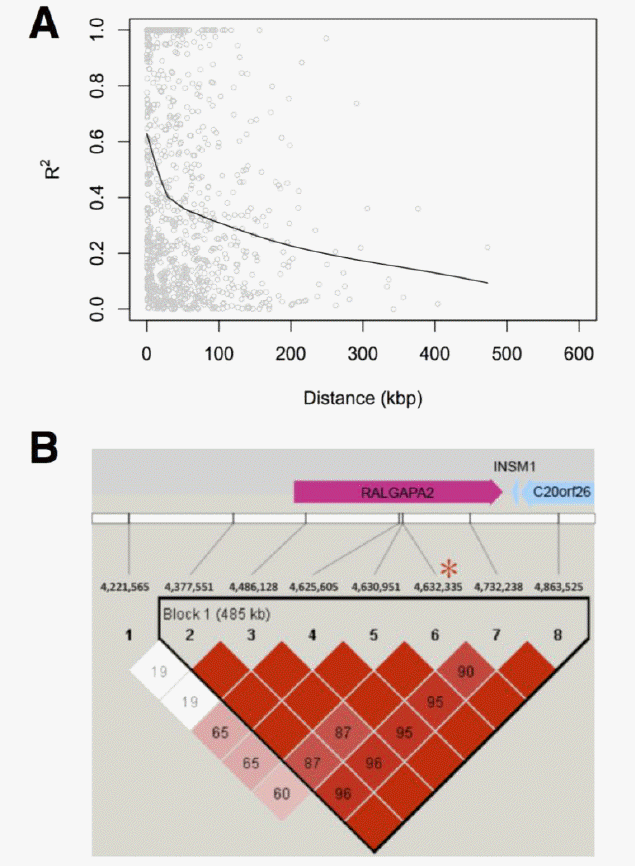
Linkage disequilibrium around potential candidate genes. The degree of linkage-disequilibrium (LD) decrease as a function of distance (A). Red asterisk depicts the position of genetic maker significantly affecting racing performance on horse chromosome 22 (B).
DISCUSSION
The population differentiation index has the power to detect selection on standing variation as well as on newly selected sites (Innan and Kim, 2008). The relative level of genetic variability around the focal locus is a measure of selection pressure. We identified putative signals of strong artificial selection in Thoroughbred genomes by combining both tests.
Previous gene-centered studies have focused on genetic mechanism related to muscle contents and energy metabolism for racing performance in Thoroughbreds. For example, MSTN is known to be associated with double-muscling phenotype (Kambadur et al., 1997; McPherron and Lee, 1997; Riquetl et al., 1997). This region has a low FST index (0.08) with low π values in both the Jeju breed (20.17) and Thoroughbred (13.40), suggesting that MSTN could have an essential role in the phenotypes of muscle, and selection has been acting on this before the domestication of horse.
Our results provide strong evidence of the genetic basis for selective breeding for racing performance in horses. We found a large gene cluster around RALGAPA2 in the Thoroughbred genomes. The RalGAPs, α1, and α2, are critical for efficient termination of Ral activation induced by extracellular stimuli that recombinant RalGAP1 accelerates the guanosine triphosphate hydrolysis rate of RalA by 280,000-fold (Suzuki et al., 2000). Genes within the cluster have a similar function. For example, like RALGAPA2, pallid (PLDN), and Ras and Rab interactor 2 (RIN2) genes encode protein for cellular vehicles or membrane trafficking. 5′-3′ Exoribonuclease 2 (XRN2), RIN2, crooked neck pre-mRNA splicing factor 1 (CRNKL1), and PLDN genes are associated with cellular component organization. Insulinoma-associated 1 (INSM1) also plays an essential role in neuronal phenotype in vertebrate hindbrain (Jacob et al., 2009). Therefore, this cluster appears to have an important molecular role in controlling the function of signal trafficking, reflecting artificial selection processes driving them into a tight gene cluster for efficacy of the associated function.
In this study, based on association analysis within the context of population genomics study, we suggest that the novel candidate genes for racing performance can potentially illuminate previously unsuspected biological mechanisms. Therefore, the principal challenge of multifactorial traits may lay not only in detecting quantitative trait loci, but also in identifying targets of selection. Further functional studies are needed to investigate the downstream targets of domesticated genes affecting racing performance.
ACKNOWLEDGMENTS
This work was supported by KRA for the project of Thoroughbred horse (No. 0569-20110008). We thank Profs. Rasmus Nielsen (UC, Berkeley) and Yuseob Kim (Ewha Womans University) for critical reading and comments on the manuscript.