Imputation Accuracy from Low to Moderate Density Single Nucleotide Polymorphism Chips in a Thai Multibreed Dairy Cattle Population
Article information
Abstract
The objective of this study was to investigate the accuracy of imputation from low density (LDC) to moderate density SNP chips (MDC) in a Thai Holstein-Other multibreed dairy cattle population. Dairy cattle with complete pedigree information (n = 1,244) from 145 dairy farms were genotyped with GeneSeek GGP20K (n = 570), GGP26K (n = 540) and GGP80K (n = 134) chips. After checking for single nucleotide polymorphism (SNP) quality, 17,779 SNP markers in common between the GGP20K, GGP26K, and GGP80K were used to represent MDC. Animals were divided into two groups, a reference group (n = 912) and a test group (n = 332). The SNP markers chosen for the test group were those located in positions corresponding to GeneSeek GGP9K (n = 7,652). The LDC to MDC genotype imputation was carried out using three different software packages, namely Beagle 3.3 (population-based algorithm), FImpute 2.2 (combined family- and population-based algorithms) and Findhap 4 (combined family- and population-based algorithms). Imputation accuracies within and across chromosomes were calculated as ratios of correctly imputed SNP markers to overall imputed SNP markers. Imputation accuracy for the three software packages ranged from 76.79% to 93.94%. FImpute had higher imputation accuracy (93.94%) than Findhap (84.64%) and Beagle (76.79%). Imputation accuracies were similar and consistent across chromosomes for FImpute, but not for Findhap and Beagle. Most chromosomes that showed either high (73%) or low (80%) imputation accuracies were the same chromosomes that had above and below average linkage disequilibrium (LD; defined here as the correlation between pairs of adjacent SNP within chromosomes less than or equal to 1 Mb apart). Results indicated that FImpute was more suitable than Findhap and Beagle for genotype imputation in this Thai multibreed population. Perhaps additional increments in imputation accuracy could be achieved by increasing the completeness of pedigree information.
INTRODUCTION
Genomic information has been widely used in livestock research and has come to play an important role in the characterization and evaluation of dairy cattle. Genomic selection utilizes genomic information to increase the rate of genetic progress in dairy populations (VanRaden et al., 2009; de Roos et al., 2011). Using high density chips to obtain genomic information will increase the effectiveness of genomic selection (VanRaden et al., 2011; Mulder et al., 2012), but genotyping costs may be prohibitively high for most dairy producers. Thus, genotyping companies have produced genotyping chips of lower densities and lower cost to increase their affordability and their use by dairy producers. Genotypic information from lower density chips is subsequently imputed to a higher density chip before using it for animal genomic evaluation. This approach has drastically reduced genotyping costs compared to using only high density chips. In dairy cattle, genotype imputation is regularly utilized in many countries, and several software packages have been developed for this purpose. These software packages have yielded imputation accuracies ranging from 81% to 97% from low to moderate densities (Druet et al., 2010; Johnston et al., 2011; Ma et al., 2013; Weng et al., 2013), and from 84% to 99% from low to high densities in dairy cattle populations (Ma et al., 2013; Larmer et al., 2014; Pryce et al., 2014; He et al., 2015).
Dairy cattle have been routinely evaluated in Thailand with mixed model procedures that use phenotypic and pedigree information since the late 1990’s (DPO, 1997; Koonawootrittriron et al., 2015). In April 2012, the National Science and Technology Development Agency of Thailand approved a project to develop a national genomic-polygenic evaluation system (Koonawootrittriron et al., 2012). The aim of this project is to develop and implement genomic-polygenic evaluation strategies suitable for the Holstein-Other multibreed dairy cattle population in Thailand. This population was created using a grading-up mating strategy towards Holstein starting from animals of various breeds (Brahman, Jersey, Brown Swiss, Red Dane, Red Sindhi, Sahiwal and Thai Native). Currently, 93% of the population is at least 75% Holstein. Budgetary restrictions determined animals in the Thai Holstein-Other multibreed population to be genotyped with high, medium, and low density genotyping chips. This created the need to identify a program capable of imputing genotypes with high accuracy in the Thai multibreed population. However, imputation algorithms and software packages have been primarily tested in purebred dairy populations, but animals in the Thai multibreed population, although mostly Holstein, contained fractions of up to 7 other breeds (average 3 breeds per animal; Koonawootrittriron et al., 2009; Ritsawai et al., 2014). Further, research has shown that the accuracy of imputation depends on population structure as well as the algorithms used (Johnston et al., 2011; Larmer et al., 2014), and that accuracy of imputation will affect the reliability of genomic predictions (Khatkar et al., 2012; Mulder et al., 2012). Thus, the objective of this research was evaluate the accuracy of three population-based and combined family- and population-based software programs to impute single nucleotide polymorphism (SNP) markers to identify the most appropriate for the Thai Holstein-Other multibreed dairy cattle population.
MATERIALS AND METHODS
Animals and genotypes
A total of 1,244 animals from the Thai multibreed dairy population (84 sires and 1,160 cows) were used in this study. All sires and 50 highly related cows were genotyped with GeneSeek Genomic Profiler 80K chip (GGP80K; GeneSeek, Lincoln, NE, USA). The remaining cows were genotyped with GeneSeek Genomic Profiler 20K (GGP20K; n = 570 cows) and GeneSeek Genomic Profiler 26K (GGP26K; n = 540 cows). Sires were born between 1993 and 2009 and they were widely used for artificial insemination in the population. Cows were from 145 farms, born between 2000 and 2011, and had their first lactation between 2003 and 2014. Nearly all animals (97%) were crossbred with fractions of Holstein (H) and fractions of up to 7 other breeds (Brahman, Jersey, Brown Swiss, Red Dane, Red Sindhi, Sahiwal, and Thai Native). Ninety three percent of all animals, 96% of all sires, and 86% of all dams were between 75% and 100% Holstein. These high Holstein percentages in animals, sires, and dams were the result of the Holstein upgrading program used to create the Thai Holstein-Other multibreed dairy cattle population.
All SNP markers that were not located on autosomes were eliminated. The numbers of autosomal SNP markers per chip were 74,672 for the GGP80K, 24,572 for the GGP26K, and 18,593 for the GGP20K. To assess imputation accuracy, only SNP markers in common (name, chromosome, and position) among the GGP80K, GGP26K, and GGP20K chips were used. Unfortunately, the number of autosomal SNP markers in common among these three chips was only 8,671. However, there were 1,110 animals genotyped with GGP26K and the GGP20K that had 18,129 autosomal SNP markers in common. Therefore, 9,458 autosomal SNP markers not present in the GGP80K were imputed for the 134 animals genotyped with GGP80K based on 18,129 reference SNP markers from 1,110 cows genotyped with GGP26K and GGP20K using the combined family- and population-based option of FImpute 2.2 (Sargolzaei et al., 2014). This was done to increase the family ties among the remaining 1,110 cows in the dataset (animals genotyped with GGP80K were highly related to other animals in the population), which would help increase imputation accuracy. Subsequently, call rates and minor allele frequencies (MAF) were recalculated, and SNP markers with a MAF lower than 0.01 or a call rate lower than 0.9 were eliminated. After these quality checks, 17,779 SNP markers were kept to represent the moderate density chip (MDC).
The set of 1,244 genotyped animals were the progeny of 331 sires and 1,067 dams. Of these 1,244 animals, 1,133 had both the sire and dam identified, 108 had the sire identified only, and 3 had neither the sire nor the dam identified. To evaluate the imputation accuracy, the set of 1,244 animals were sorted by year of birth and assigned to two groups: a reference group (animals born before 2010; n = 912) and a test group (animals born in or after 2010; n = 332). All SNP markers (n = 17,779) were considered for the reference group, whereas animals in the test group were assumed to have been genotyped only for the subset of SNP contained in the GeneSeek Genomic Profiler 9K chip (LDC; n = 7,652 SNP markers).
Imputation algorithm and accuracy
Genomic imputation was performed using three different software packages, namely Beagle 3.3 (Browning and Browning, 2009), FImpute 2.2, and Findhap 4 (VanRaden and Sun, 2014). These three software packages were chosen because they had been found to have high imputation accuracies in cattle populations (Johnston et al., 2011; Sun et al., 2012; Ma et al., 2013).
Beagle relies on a population-based algorithm that assumes all animals to be unrelated. It uses a hidden Markov model for constructing haplotypes and imputing unknown genotypes (Browning and Browning, 2009). FImpute and Findhap use a combined family- and population-based algorithms for phasing haplotypes and imputing unknown genotypes (VanRaden et al., 2011; Sargolzaei et al., 2014). FImpute uses pedigree information first to impute unknown genotypes using known relationships among animals. Then, it uses a population-based algorithm (overlapping sliding windows) to find shared haplotype segments, under the assumption that all animals are related to each other. This process finds from long to short shared haplotypes to capture as many as haplotypes from close to far relatives. Shared haplotypes are used to impute unknown genotypes (Sargolzaei et al., 2014). Findhap divides each chromosome into short segments before constructing reference haplotypes. Reference haplotypes are assigned to a haplotype library and sorted from most to least frequently found. Haplotypes from target animals are searched to match haplotypes in the library and then matched haplotypes are used to fill unknowns. Pedigree information is utilized to trace matching haplotypes for animals with known relationships (VanRaden et al., 2011).
The three imputation programs were run using the recommended default parameter settings. After completing the imputation process, imputed genotypes were compared to the actual genotypes. Imputation accuracies was assessed within and across chromosomes using the concordance rate (Piccoli et al., 2014) computed as the ratio of correctly imputed SNP markers to overall imputed SNP markers across the 332 animals in the test group. Missing SNP markers in all animals were excluded from accuracy computations.
Relationship between linkage disequilibrium and imputation accuracy
Linkage disequilibria among SNP genotypes in the reference group were computed as correlation coefficients (r2) between pairs of loci within chromosomes that were less than or equal to 1 Mb apart. The r2 values were estimated using Haploview 4.2 (Barrett et al., 2005). The average linkage disequilibrium (LD) of an SNP was calculated as the average of all LD values between this SNP and all SNP within 1 Mb from it. Average LD of imputed SNP markers was plotted against their imputation accuracies to examine the relationship between LD and imputation accuracy.
RESULTS AND DISCUSSION
Imputation accuracy
Table 1 shows imputation accuracies from LDC to MDC for the three software packages. FImpute had the highest imputation accuracy (93.94%), Findhap was second (84.64%), and the least accurate was Beagle (76.79%). FImpute and Findhap utilize pedigree information for constructing haplotypes and imputing unknown genotypes, whereas Beagle does not require this information (Browning and Browning, 2009; VanRaden et al., 2011; Sargolzaei et al., 2014). Thus, results here showed that pedigree information was useful for improving imputation accuracy in this Thai multibreed dairy population. This agreed with the finding that when imputing from low to MDCs, pedigree information is important to achieve high imputation accuracy, especially when reference populations are small (Sargolzaei et al., 2014). Pedigree information may help to more accurately trace shared haplotype segments between reference and test animals before they are used to impute unknown genotypes (Kong et al., 2008; Hayes et al., 2012). However, the importance of pedigree information may decrease when using a large reference population with high-density genotypic information (Ma et al., 2013; Larmer et al., 2014; Sargolzaei et al., 2014).

Imputation accuracy using Beagle, FImpute, and Findhap
The higher imputation accuracies obtained here for the combined family- and population-based algorithms than for population-based algorithm were in agreement with previous studies in various dairy cattle populations. FImpute and Findhap outperformed Beagle in Norwegian Holstein (Johnston et al., 2011) and Chinese Holstein (He et al., 2015). FImpute outperformed Beagle in Holstein (Sargolzaei et al., 2014) and Guernsey and Ayrshire populations (Larmer et al., 2014). Conversely, Beagle outperformed FImpute and Findhap in Swedish and Finnish Red cattle (Ma et al., 2013). Beagle accounts for genomic family relationships between reference and test animals using shared haplotype segments, thus the longer the shared haplotype segment the stronger the genomic relationship between animals and the higher the imputation accuracy (Browning and Browning, 2009; Ma et al., 2013). Conversely, pedigree errors decrease the accuracy of family-based algorithm (Sargolzaei et al., 2014). Thus, perhaps strong genomic relationships or pedigree errors may have contributed to the advantage of Beagle over FImpute in the Swedish and Finnish Red cattle populations.
This study showed that combined family- and population-based algorithms (FImpute and Findhap) performed acceptably in the Thai dairy cattle population (over 80% correctly imputed SNP). However, imputation accuracy was 9.3% higher for FImpute than Findhap. This superiority of FImpute over Findhap may have been due to a more effective utilization of short haplotype segments to account for genomic relationships among animals in the reference and test subpopulations. The Thai dairy population is multibreed, thus shared haplotypes among animals are likely to be short (Ventura et al., 2014). FImpute uses an overlapping sliding window algorithm to more efficiently capture short shared haplotypes (Sargolzaei et al., 2014). This algorithm likely helped FImpute compute genomic relationships more accurately resulting in higher imputation accuracies than Findhap in this population. Lastly, an advantage of both FImpute and Findhap over Beagle is that these two programs can impute ungenotyped animals if strong relationships among genotyped and non-genotyped animals existed (VanRaden et al., 2011; Sargolzaei et al., 2014). Imputing ungenotyped animals would help increase the accuracy of genomic evaluation in a population.
Imputation accuracies obtained here with the three programs used in this population were lower than accuracies found for other dairy populations. The accuracy reported for imputation from low to MDCs ranged from 92.0% to 96.5% for Beagle (Johnston et al., 2011; Ma et al., 2013; Weng et al., 2013), 95.0% to 96.8% for FImpute (Johnston et al., 2011; Ma et al., 2013), and 89.0% to 95.9% for Findhap (Johnston et al., 2011; Ma et al., 2013). Shared haplotype segments among animals in multibreed populations are likely to be shorter than in purebred populations (Ventura et al., 2014; Fu et al., 2015). As previously explained, the Thai multibreed population is composed of Holstein and seven other Bos taurus and Bos indicus breeds. Thus, the lower accuracies obtained here may have been due to lower number and shorter shared haplotype segments among animals in the reference and test populations than populations used to assess imputation accuracy elsewhere (mostly animals of a single breed). The small reference population size could be one other reason for this low accuracy. Several studies have shown that increasing the number of animals in the reference population can increase imputation accuracy (Druet et al., 2010; Zhang and Druet, 2010; Khatkar et al., 2012). Although somewhat lower than in purebred populations, the relatively high imputation accuracy of FImpute here suggested that this would be the imputation program of choice for the Thai multibreed population.
Imputation accuracy was also computed within the 29 autosomal chromosomes (Figure 1). Accuracies were similar and consistent across chromosomes for FImpute, but not for Findhap and Beagle. Among the 29 chromosomes, imputation accuracies ranged from 92.02% to 95.21% for FImpute, 78.70% to 87.94% for Findhap, and 71.82% to 84.09% for Beagle. Thus, FImpute showed higher and more consistent levels of imputation accuracy across chromosomes than Findhap and Beagle. The higher within-chromosome and whole-genome imputation accuracies obtained with FImpute indicated that this program was the most suitable of the three programs evaluated here for the Thai multibreed population. Further, the accuracy levels obtained with FImpute suggested that it could help improve the accuracy of genomic evaluation in Thai dairy cattle.
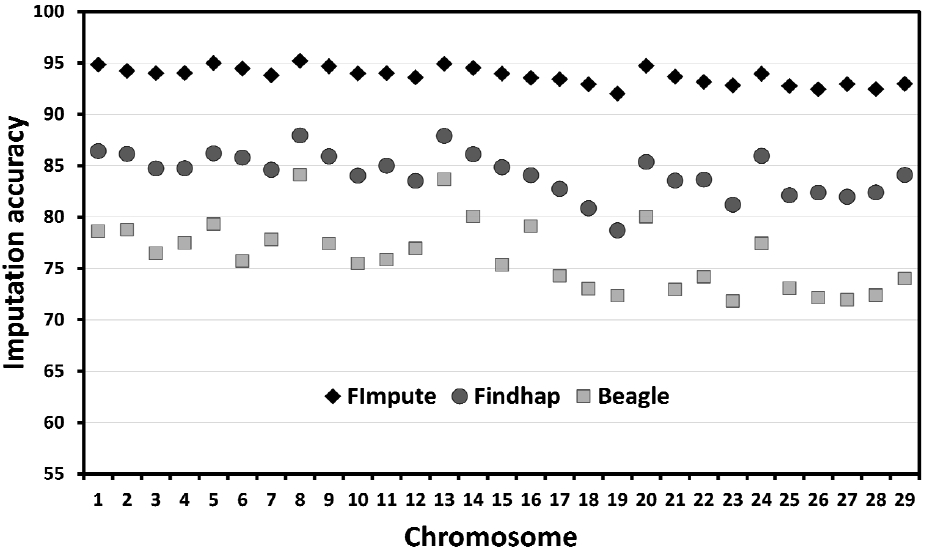
Imputation accuracy within the 29 autosomal chromosomes using Beagle, FImpute, and Findhap.
Relationship between linkage disequilibrium and imputation accuracy
The average LD (r2) across 29 chromosomes estimated using MDC genotypic information (17,779 SNP) from animals in the reference group was 0.069. The highest average LD were obtained in chromosome 5 (r2 = 0.124), and the lowest average LD occurred in chromosome 28 (r2 = 0.047; Figure 2). The distribution of average LD for individual SNP (Figure 3) shows that most SNP markers (84%) had low levels of average LD (smaller than 0.10).

Average linkage disequilibrium (correlation coefficient; r2) between adjacent single nucleotide polymorphism makers separated by at most 1 Mb within each autosome.
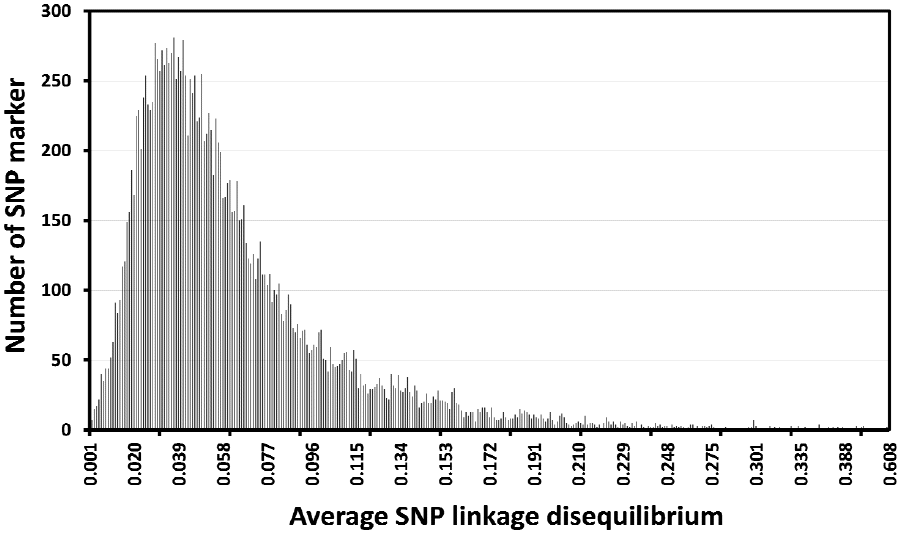
Distribution of average SNP linkage disequilibria (LD; correlation coefficient; r2) for SNP within 1 Mb of each other. SNP, single nucleotide polymorphism.
Imputation accuracy was found to be affected by the level of LD (Hickey et al., 2012; Pimentel et al., 2013). The higher the LD population has the higher the imputation accuracy. Results here agreed with these studies. Most chromosomes (73%) that had higher than average imputation accuracy (chromosomes 1, 2, 3, 4, 5, 7, 8, 9, 13, 14, 24) also had higher than average LD (chromosomes 1, 2, 3, 5, 7, 9, 13, 14). Conversely, most chromosomes (80%) that had lower than average imputation accuracy (chromosomes 17, 18, 19, 22, 23, 25, 26, 27, 28, 29) also had lower than average LD (chromosomes 17, 18, 22, 25, 26, 27, 28, 29). Thus, low average LD appeared to have decreased the ability of the imputation programs used here to find shared haplotypes, resulting in lower imputation accuracies. However, many SNP markers (30% for FImpute and Findhap and 33% for Beagle) that had below average LD had above average imputation accuracies (Figure 4). Thus, discarding SNP with below average LD would likely be ineffective to increase imputation accuracy. Alternatively, increasing the number of pedigree ties between the reference and test populations as well as increasing the number of animals in the reference population would likely be more effective to improve imputation accuracy (Druet et al., 2010; Zhang and Druet, 2010; Sargolzaei et al., 2014).

Imputation accuracy by average SNP linkage disequilibrium (LD; correlation coefficient; r2) at distances between SNP lower than or equal to 1 Mb computed using Beagle, FImpute and Findhap. SNP, single nucleotide polymorphism.
CONCLUSION
Accuracy of imputation from LDC to MDC in the Thai multibreed dairy cattle population ranged from 76.79% to 93.94%. FImpute (combined family- and population-based algorithms; Accuracy = 93.94%) outperformed Findhap (combined family- and population-based algorithms; Accuracy = 84.64%) and Beagle (population-based algorithm; Accuracy = 76.79%). Imputation accuracies were similar and consistent across chromosomes for FImpute, but not for Findhap and Beagle. Increasing pedigree ties between reference and test populations and increasing size of the reference population will likely help improve imputation accuracy.
ACKNOWLEDGMENTS
The authors would like to thank the Royal Golden Jubilee Ph.D. Program (RGJ) of the Thailand Research Fund (TRF) for awarding the scholarship to the first author, the University of Florida for supporting the training of the first author as a research scholar, and the National Science and Technology Development Agency (NSTDA), Kasetsart University (KU), and the Dairy Farming Promotion Organization of Thailand (D.P.O.) for providing funding and support for this research. The authors also appreciate the Thai dairy farmers, dairy cooperatives, and dairy related organizations for their support of this investigation.
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.