





INTRODUCTION
Hair color is one of the most important factors affecting the economic value of fiber-producing animals. Hair color genes are also useful candidates for the traceability of farm animals [1]. Mouse models have been extensively used to investigate the functions of hair color-related genes. These models have received increasing attention, revealing that the fiber diameter, length, and color are determined by both genetics [2,3] and environment [4]. Hair and skin color depend on the pigments produced by melanocytes at the base of the epithelium [5]. Mammalian melanocytes produce two chemically distinct types of melanin, black/brown eumelanin and yellow/red-brown pheomelanin [6]. The quality and ratio of eumelanin to pheomelanin determine the final color of the hair and skin. Through extensive studies, the genetic basis for hair and skin color in rodents has been relatively well elucidated, with many genes involved in pigmentation being common to other species. For example, microphthalmia-associated transcription factor (MITF) plays an important role in skin color and melanoma [7] and the agouti signaling protein (ASIP) is a major regulator of mouse pigmentation [8].
Cyclin dependent kinases (CDKs) are proline-directed serine/threonine protein kinases that play important roles in cell cycle regulation [9]. However, cyclin-dependent kinase 5 (CDK5) is notable in that it does not appear to be directly involved in the cell cycle, is not activated after binding to cyclin, and does not require T-loop phosphorylation for activation [10]. An analysis of the transcriptome profile of alpaca skin with different hair colors revealed that CDK5 is a candidate gene associated with alpaca fleece quality, coat color, and fiber growth and development [11]. Cyclin-dependent kinase 5 is localized in hair follicles, with higher expression levels in animals with brown than in those with white fleece colors. Our previous study investigating the genetic components of hair color in a mouse model showed that CDK5 plays a role in determining hair color in mice and a change in hair color from black to light brown was noted [11]. To further investigate the genes or pathways involved in pigmentation regulated by CDK5, CDK5-knockdown mice were generated, and skin mRNA profiles from CDK5-knockdown mice were constructed.
This study revealed that numerous mRNA transcripts are regulated by CDK5, providing insights into its potential functions. These data will contribute to a better understanding of how hair and skin pigmentation are determined in animals of economic importance and may lead to the design of more productive or desirable breeds.
MATERIALS AND METHODS
Ethics statement
The procedures for animal housing, care, and collection of skin samples were approved by the Animal Ethics Committee (2017[050]) at Shanxi Agriculture University (Taigu, Shanxi, China; Approval number: 2023008) and performed according to the Committee guidelines.
Skin sampling and total RNA extraction
The CDK5-knockdown mouse models were prepared according to previously described instructions [12]. Three healthy wild-type and three CDK5-knockdown mice were randomly selected, and three pieces of skin (2├Ś3 cm) from their backs were collected under local anesthesia and immediately stored in liquid nitrogen. Total RNA was extracted from the samples using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturerŌĆÖs instructions. The RNA integrity was evaluated by gel electrophoresis, and the concentration was measured by absorbance at optical density (OD260/280) using a NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Library generation and sequencing
After total RNA extraction and DNase I treatment, magnetic beads bound to oligo-dT were used to isolate the mRNAs. These were mixed with a fragmentation buffer to break the mRNAs into short fragments. cDNA was synthesized from the mRNA fragment templates. Short fragments were purified and resolved using the Qiagen EB buffer (Qiagen, Hilden, Germany) for end preparation and single-nucleotide adenine addition. These fragments were then ligated to adapters and suitably sized fragments were selected for polymerase chain reaction (PCR) amplification using agarose gel electrophoresis. During these quality control steps, an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and an ABI StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) were used for mRNA quantification and qualification. Finally, the library was sequenced using an Illumina HiSeq 2000 (Illumina, San Diego, CA, USA) to generate raw reads, which were filtered into clean reads by removing adaptors and low-quality reads, and then aligned to the reference sequences. These alignment data were used to calculate the distribution of reads on the reference genes and mapping ratios for further analyses, including gene ontology (GO) enrichment analysis, Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis, and transcription factors identification.
UniGene assembly and functional annotation
UniGene assembly was performed using the Trinity software (http://www.genomics.cn). BLASTX alignment (e-values <0.00001) between the unigenes and several protein databases (nr, Swiss-Prot, KEGG, and clusters of orthologous genes) was performed, with the best alignment results used to determine the sequence direction of the unigenes. GO functional annotation was based on the nr annotation, and Blast2GO (http://www.blast2go.com) was used to assign GO annotations. WEGO (http://wego.genomics.org.cn/cgibin/wego/index.pl) was used to perform the GO functional classifications of all unigenes.
Identification of differentially expressed genes and pathway analysis
A rigorous algorithm was used to identify genes in the skin that are differentially expressed between CDK5-knockdown and wild-type mice. The algorithm consisted of a threshold false discovery rate value of Ōēż0.001 and a reads per kilobase of transcript per million mapped reads ratio of Ōēź2. Differentially expressed genes were mapped to each term in the GO database (http://www.geneontology.org/) and the number of genes assigned to each GO term was calculated. The calculated p-values were Bonferroni corrected and corrected p-values Ōēż0.05 were determined to be significant. GO terms that fulfilled this condition were defined as significantly enriched. The differentially expressed genes was also mapped to terms on the KEGG pathway database (http://www.genome.jp/kegg/pathway.html) to reveal the biological pathways that differed between CDK5-knockdown and wild-type mice.
Validation of differential gene expression in the skins of CDK5-knockdown and wild-type mice
To validate the sequencing data, 10 genes were randomly selected from a list of differentially expressed genes using the quantitative real-time PCR (qRT-PCR). Total RNA from six samples used for RNA sequencing was used for this analysis. One microgram of DNase-treated RNA was converted to cDNA using the PrimeScript RT Reagent Kit (TaKaRa, Dalian, China). The cDNA was used for qRT-PCR quantification using mRNA-specific primers (Table 1). ╬▓-Actin was used as an endogenous control. The qRT-PCR was performed in triplicate using a Stratagene Mx3005P system (Stratagene, La Jolla, CA, USA). Each 10 ╬╝L PCR reaction volume included 5 ╬╝L of SYBR Premix Ex TaqTM II (TaKaRa, China), 0.2 ╬╝L of specific forward primer, 0.2 ╬╝L of reverse primer, 0.2 ╬╝L of ROX reference dye, 1 ╬╝L of 10-fold diluted cDNA, and 3.4 ╬╝L of water. The cycling parameters were 95┬░C for 30 s, followed by 40 cycles of 95┬░C for 5 s, 56┬░C or 58┬░C for 30 s, and 72┬░C for 15 s. Melting curve analysis was performed at each amplification step. The mRNA abundance of each target gene was quantified using the comparative threshold cycle (CT) method and normalized to ╬▓-actin [13]. Differences in the mRNA abundance of genes were determined using analysis of variance.
RESULTS
Sequencing statistics
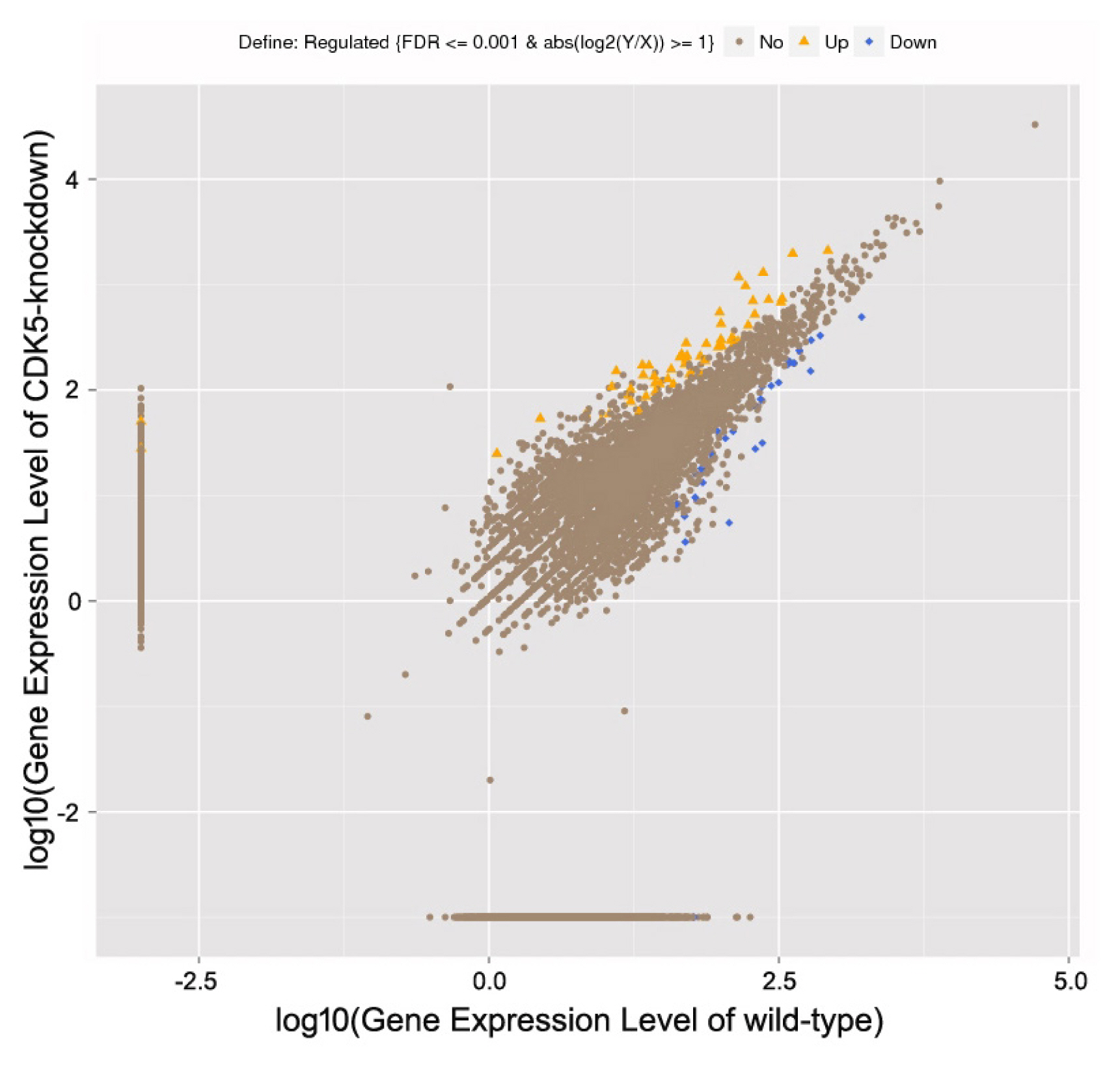
To identify the RNAs expressed in mouse skin, wild-type and CDK5-knockdown skin mRNA libraries were generated using high-throughput sequencing. A total of 98,801,386 and 96,503,448 raw reads were obtained from the wild-type and CDK5-knockdown mice, respectively. After filtering low-quality reads, 86,182,330 (87.23%) and 86,398,588 (89.53%) clean reads remained in the wild-type and CDK5-knockdown datasets, respectively. Using an algorithm based on a previously described method [14], 8,002 known genes were identified as differentially expressed between the skin of CDK5-knockdown and wild-type mice. Of these, 4,344 were downregulated (Ōēż2-fold) and 3,658 were upregulated (Ōēź2-fold) in CDK5-knockdown mice compared to wild-type (Figure 1; Supplementary Table S1).
Gene ontology analysis of genes expressed in the skin of CDK5-knockdown versus wild-type mice
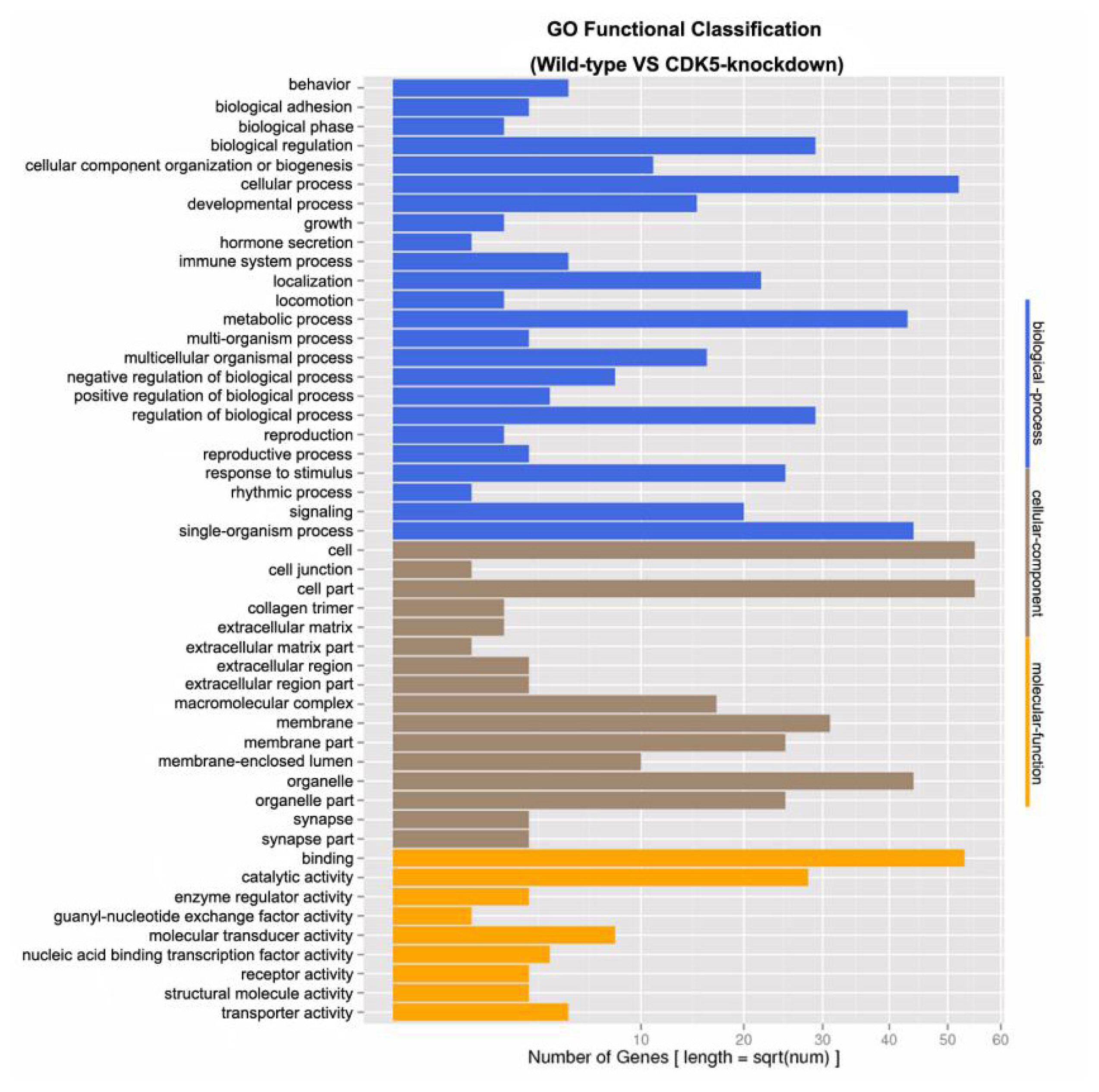
Genes differentially expressed between the skin of CDK5-knockdown and wild-type mice were grouped into 49 classes based on their putative functions. For subsequent GO analysis, the differentially expressed genes were characterized at the biological process (48.98%), cellular component (32.65%), and molecular function (18.37%) levels (Figure 2). A total of 318 unknown genes were also identified as differentially expressed, of which 171 were downregulated (Ōēż2-fold) and 147 were upregulated (Ōēź2-fold) in the skin of CDK5-knockdown mice compared to the skin of wild-type mice (Supplementary Table S2).
Differential expression of known genes related to hair color formation
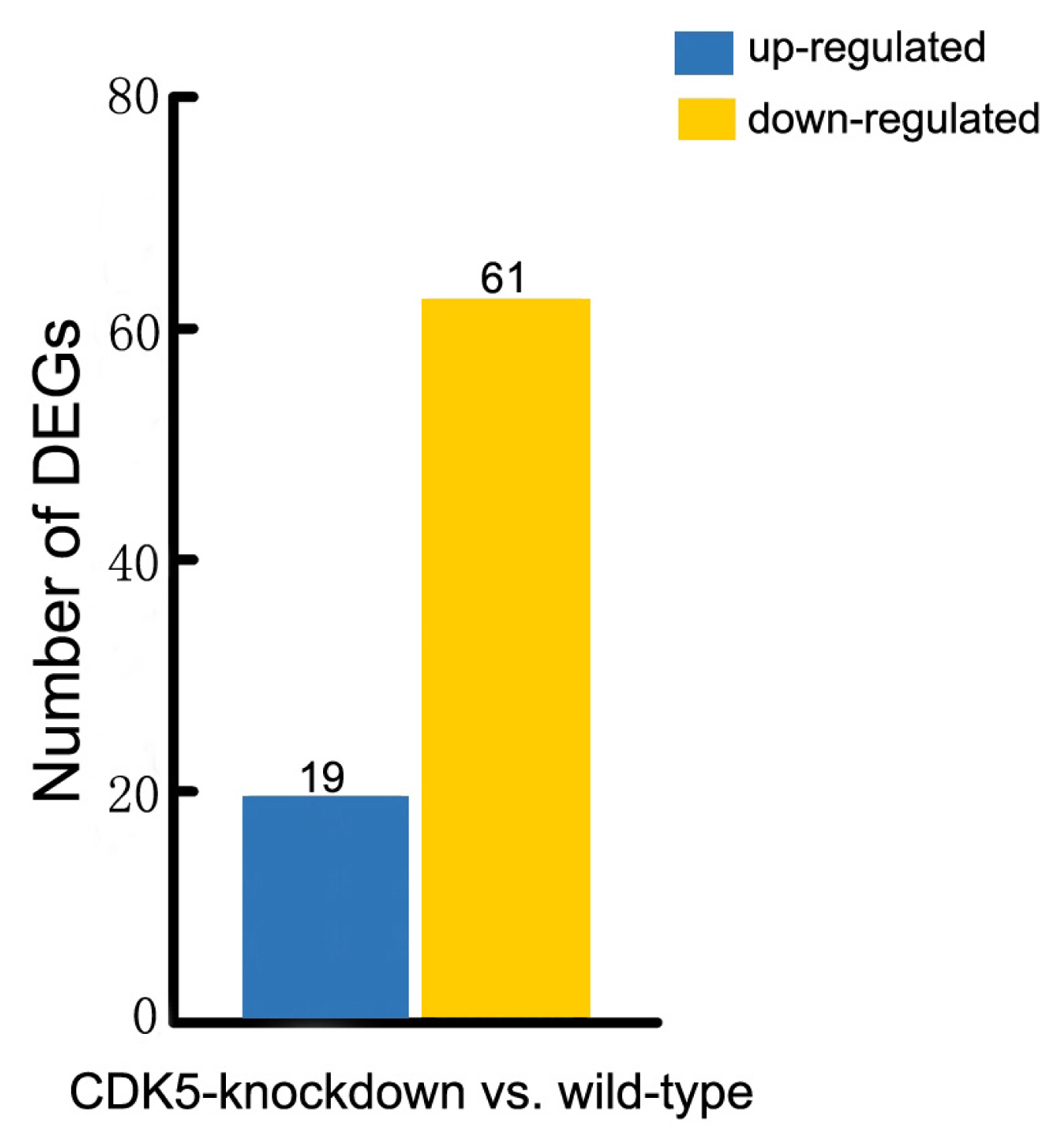
More than 150 mutations affecting hair and skin pigmentation have been identified in mice. They are well dispersed throughout the mouse genome and are found at more than 50 distinct genetic loci [15]. Known hair color genes are typically classified into six functional groups: melanocyte development, melanosome components and precursors, melanosome construction and protein routing, melanosome transport, eumelanin and pheomelanin production, and other wider systemic effects [14]. In this study we found that 80 of these hair color genes are expressed in mouse skin. Our analysis revealed that 61 of these genes were expressed at lower levels in the skin of CDK5-knockdown mice than in wild-type mice, and 19 genes were expressed at higher levels (Figure 3; Supplementary Table S3). We also found that all the genes involved in encoding the components of melanosomes and their precursors were expressed at lower levels in the skin of CDK5-knockdown mice than in the skin of wild-type mice (Supplementary Table S4). Most genes involved in the production of eumelanin and pheomelanin also showed lower expression levels in the CDK5-knockdown mice. Among the hair color genes with lower expression in the skin of CDK5-knockdown mice, tyrosinase-related protein 1 (TYRP1) showed the largest decrease in expression between CDK5-knockdown mice and wild-type mice, followed by paired box3 (PAX3), dopachrome tautomerase (DCT), adenomatous polyposis coli (Apc), and lysosomal trafficking regulator (Lyst) genes (Table 2).
Kyoto encyclopedia of genes and genomes pathway analysis
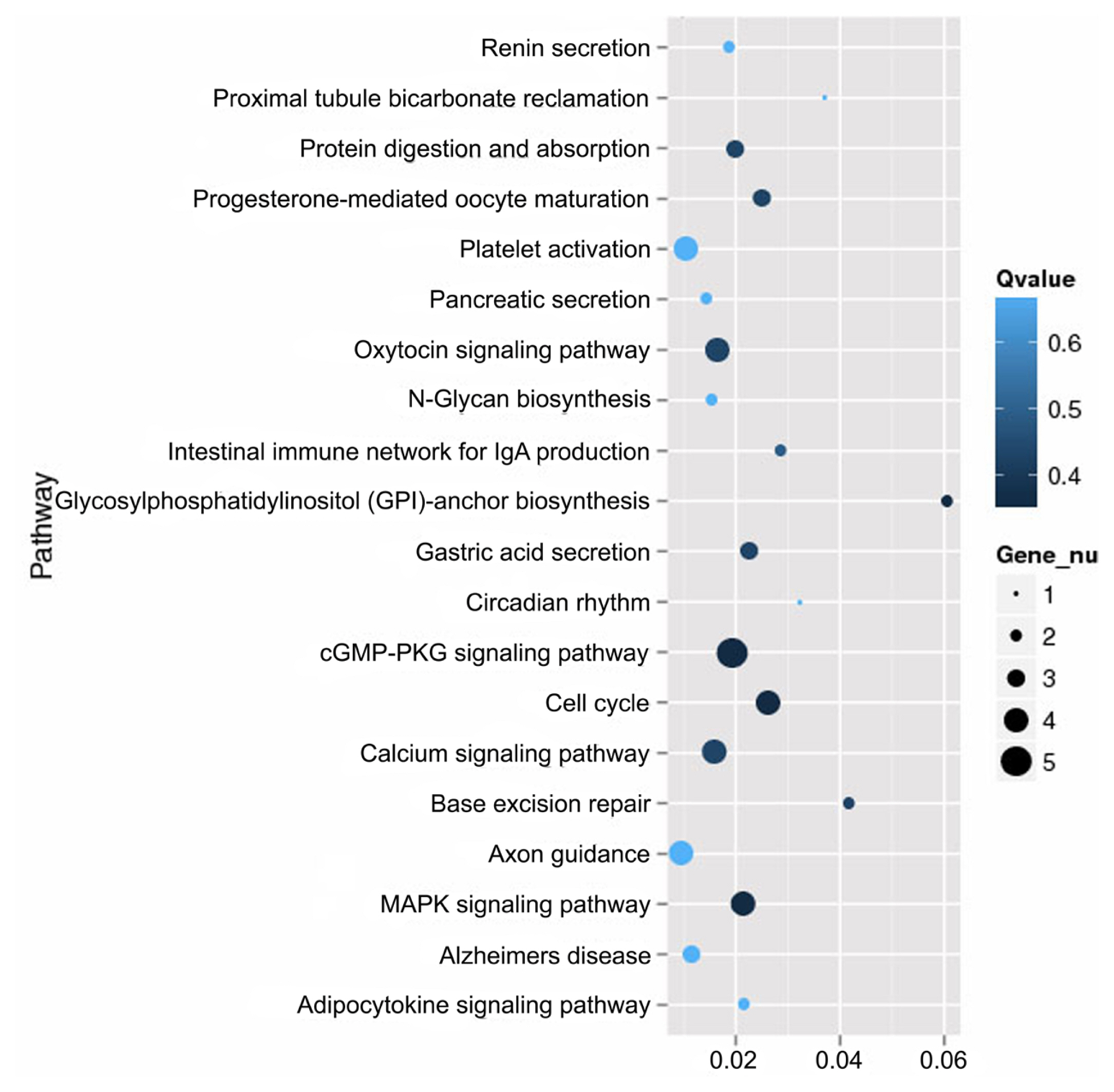
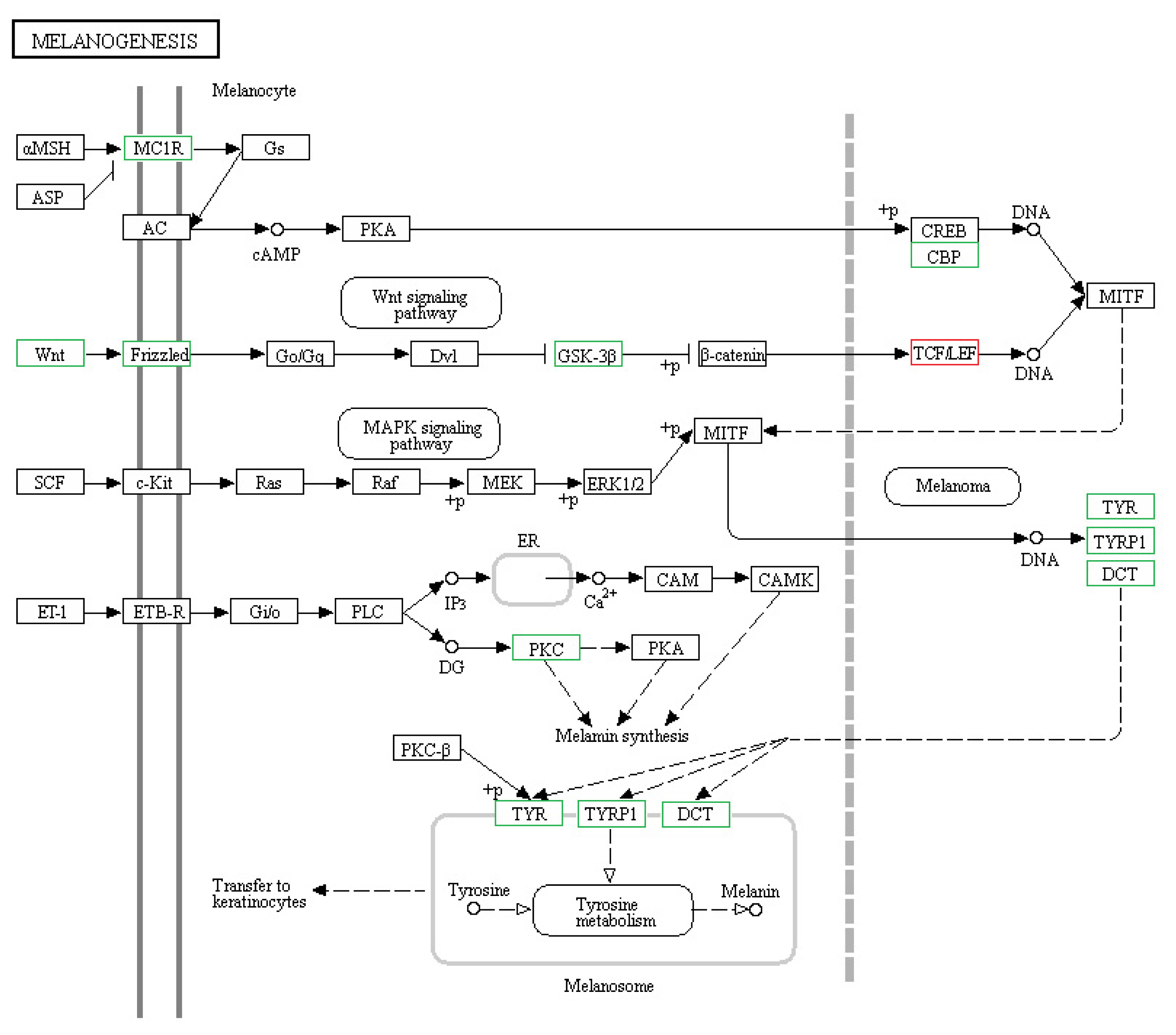
Of the 8,004 genes that were differentially expressed between the skin of CDK5-knockdown mice and wild-type mice, 234 had specific KEGG annotations. The top 20 pathways that were most highly expressed in the skin of mice included the cGMP-PKG signaling pathway, calcium signaling pathway, and MAPK signaling pathway (Figure 4). The genes identified in the mouse skin transcriptome related to pigmentation and melanogenesis and their relative level of differential expression between the CDK5-knockdown and wild-type mice are shown in Table 3 and Figure 5.
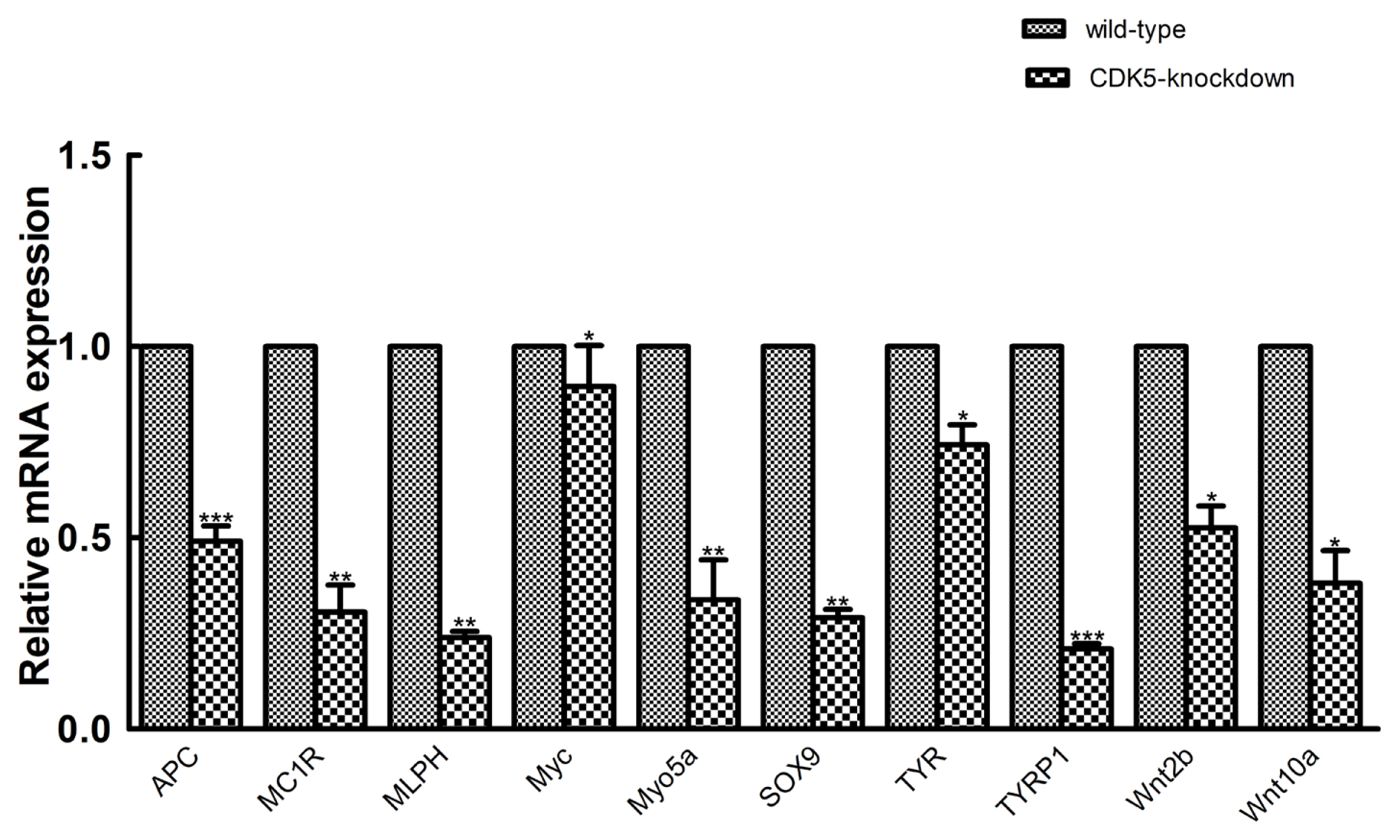
Validation of sequencing data by quantitative real-time polymerase chain reaction
To validate the transcriptome sequencing results, we randomly selected 10 genes from a population of genes known to be involved in hair color for validation using qRT-PCR. Transcriptome sequencing analysis revealed that these genes were also differentially expressed between the CDK5-knockdown and wild-type mice. The results of qRT-PCR analysis showed that the mRNA expression of the selected genes was consistent with the transcriptome sequencing data (Figure 6). Among the differentially expressed genes, TYRP1 showed the greatest difference in expression between CDK5-knockdown and wild-type mice, again supporting the sequencing data.
DISCUSSION
Mammalian hair color exhibits a wide range of shades that are dictated by the production of melanin by melanocytes (melanogenesis). These are specialized cells that synthesize two primary melanins (eumelanin and pheomelanin) [16]. The quality of these melanins and the ratio of eumelanin to pheomelanin determine the final hair and skin colors [17]. During the development of skin and hair pigmentation, precisely coordinated mechanisms play a role in regulating the various processes that lead to the final color.
This study identified several genes known to be involved in hair color, such as SOX9. Sox (SRY type HMG box) proteins are transcription factors that belong to the HMG box superfamily of DNA-binding proteins and play a key role during development. SOX9 belongs to the SOX-E subgroup, which includes SOX8, SOX9, and SOX10. SOX9 directly binds and regulates the expression of MITF [18], which subsequently regulates the transcription of three major pigmentation enzyme genes, tyrosinase (TYR), tyrosine related protein-1 (TYRP1), and tyrosine related protein-2 (TYRP2; also known as DCT) [19]. When levels of MITF reach a certain threshold, repression is removed, allowing activation of TYRP2 transcription in the presence of ╬▓-catenin, leading to melanocyte maturation [20]. Ectopic SOX9 expression in the neural crest (NC) is sufficient to promote melanocytic differentiation, which suggests a role for SOX9 in melanocytic development [21]. In summary, SOX9 appears to be an important CDK5-regulated transcription factor that is involved in the differences in hair color observed in CDK5-knockdown mice. We also found some important transcription factors and signaling pathways in the CDK5-knockdown mice, such as PAX3 [11] and the MAPK signaling pathway.
Among the differentially expressed hair color genes identified, TYRP1 showed the largest decrease in expression in the skins of CDK5-knockdown, compared to that of wild-type mice. TYRP1, a type I membrane-bound protein, is specifically expressed in melanocytes and involved in melanin production. TYRP1 is mostly expressed in the retinal pigment epithelium [22]. Melanocytes, which are derived from the NC, can be classified into two groups: cutaneous/classical and noncutaneous/nonclassical melanocytes. TYRP1 is also highly expressed in tumors derived from melanocytes such as in cutaneous and uveal melanomas. The three members of the tyrosinase family (TYR, TYRP2(DCT), and TYRP1) are regulated by the same transcription factor, MITF. In melanocytes, TYRP2 (DCT) catalyzes the rapid conversion of dopaquinone to 5,6-dihydroxyindole-2-carboxylic acid (DHICA), and then TYRP1 catalyzes the oxidation of DHICA to eumelanin [23]. TYRP1 also contributes to melanosomal structure and maturation [24]. Mutation of TYRP1, which leads to the mouse ŌĆ£lightŌĆØ phenotype (hairs pigmented only at their tips), has been associated with disrupted melanosomal structures. Therefore, it is likely that TYRP1 is an important multifunctional melanogenic protein that indirectly contributes to the regulation of melanin production in CDK5-knockdown mice.
Additionally, KEGG pathway analysis revealed that many differentially regulated pathways are involved in pigmentation or melanogenesis. These include the calcium, cGMP-PKG, and MAPK signaling pathways. In the electron transfer system, calcium plays an important role in both melanocyte function and survival [25]. An increase in free intracellular Ca2+ levels may be important for stimulating melanogenesis [26]. Previous studies have suggested that increasing Ca2+ level inhibits basal melanogenesis, as increasing the amount of Ca2+ in B16 [27] and human melanocytes [28] resulted in decreased tyrosinase activity, and hence a reduced basal melanin content.
In summary, our study revealed differentially expressed genes and pathways in the skin of CDK5-knockdown mice, indicating the physiological functions of CDK5 in melanogenesis. This finding that the CDK5 regulation of melanin production potentially occurs via the potential calcium signaling pathway and TYRP1 will contribute to the understanding of animal hair and skin color development.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement1
Supplement1 Print
Print







